- Awareness. The trouble with filler words is that we are completely unaware that we’re using them. My filler word when I first started training was, “okay.” I probably said “okay” fifteen times every ten minutes. During my first week with ExecuTrain, I was video taped and told to watch for the “okay’s.” I was dumbfounded! I couldn’t believe how many times I used that word.
- Practice and persistence. Once I became aware of the problem, I started hearing it myself while it was happening. For the next few weeks, when I was teaching a class, I would make a mark on a sheet of paper every time I heard myself say, “okay.” Just doing that immediately reduced the number of times I used it and eventually eliminated it all together. Then I started marking other filler words until I had them all eliminated.
- Silence. Instead of saying Um, say nothing. It’s okay to take a moment to stop and think. What you don’t realize is how little time it takes your brain to process all the information it needs and come back with an intelligent response. When I was video taped in college during a teaching presentation, I had what I thought were several long pauses. I was embarrassed and thought my professor would take points off for them. When I watched the video, I was amazed at how little time actually passed while I was thinking of an appropriate answer to a question posed by someone from my audience.
- Substitution. If you need to look something up, say so. If you don’t know the answer to a question off the top of your head, instead of saying, “Uh,” say, “I’m not sure; let me look that up for you.” No one expects you to know everything about everything. In fact, most of us get annoyed with people who pretend to know everything!
Thursday, September 30, 2010
Filler words ('Uhhmmm') when presenting
Thesis and Dissertations
http://ir.lib.sfu.ca/handle/1892/112
https://circle.ubc.ca/handle/2429/24
http://resources.library.ubc.ca/841/
https://circle.ubc.ca/handle/2429/24
http://resources.library.ubc.ca/841/
Installing R and JGR in Ubuntu Intrepid
Installing R and JGR in Ubuntu Intrepid
Here’s an update to my previous instructions on getting R set up and working in Ubuntu. These work for Ubuntu Intrepid, but won’t work for Ubuntu Jaunty (due out next month) until you see “jaunty” in this list.R is a free, open source software package for performing statistical analyses. It is an alternative to commercial tools such as SPSS, SAS, and S. I recommend using the RSeek search engine to search for help and 3rd party libraries.
To install R, open the Terminal console and run these commands:
gpg --keyserver subkeys.pgp.net --recv-key E2A11821Then open up your apt-get sources list for editing:
gpg -a --export E2A11821 | sudo apt-key add -
sudo gedit /etc/apt/sources.listAdd this line to the bottom of the sources.list file:
deb http://rh-mirror.linux.iastate.edu/CRAN/bin/linux/ubuntu intrepid/Note the trailing slash at the end of “intrepid/”. Also you can replace “rh-mirror.linux.iastate.edu” with another mirror server of your choice. Save the file and go back to the Terminal.
Now type this in the terminal to update apt-get’s database before you install R:
sudo apt-get updateInstall R with this command:
sudo apt-get install r-base r-base-dev r-recommendedThen run “R” to start R, and “q()” to quit it.
To get JGR, the Java GUI interface working (requires you have java installed, see the sun-java6 package), run this on the command line:
sudo R CMD javareconf
Wednesday, September 29, 2010
Riboswitches
http://en.wikipedia.org/wiki/Riboswitch
Riboswitches are often conceptually divided into two parts: an aptamer and an expression platform. The aptamer directly binds the small molecule, and the expression platform undergoes structural changes in response to the changes in the aptamer. The expression platform is what regulates gene expression.
Expression platforms typically turn off gene expression in response to the small molecule, but some turn it on. The following riboswitch mechanisms have been experimentally demonstrated.
http://www.ploscompbiol.org/article/info:doi%2F10.1371%2Fjournal.pcbi.1000823
Transat—A Method for Detecting the Conserved Helices of Functional RNA Structures, Including Transient, Pseudo-Knotted and Alternative Structures
Nicholas J. P. Wiebe, Irmtraud M. Meyer
One sequence can have multiple RNA secondary structures, transient structures
Riboswitches are often conceptually divided into two parts: an aptamer and an expression platform. The aptamer directly binds the small molecule, and the expression platform undergoes structural changes in response to the changes in the aptamer. The expression platform is what regulates gene expression.
Expression platforms typically turn off gene expression in response to the small molecule, but some turn it on. The following riboswitch mechanisms have been experimentally demonstrated.
http://www.ploscompbiol.org/article/info:doi%2F10.1371%2Fjournal.pcbi.1000823
Transat—A Method for Detecting the Conserved Helices of Functional RNA Structures, Including Transient, Pseudo-Knotted and Alternative Structures
Nicholas J. P. Wiebe, Irmtraud M. Meyer
- predict evolutionarily conserved helices that are likely to play a role in the co-transcriptional formation of the functional RNA structure(s) in vivo
- do not require a detailed knowledge of the in vivo environment, e.g. transcriptional speed, ion concentrations, interaction partners etc., and keep the number of free parameters and assumptions incorporated into the method to a minimum
- estimate reliability values for all predictions
- present a comprehensive performance evaluation
- have a performance which is robust with respect to sequence length
Motivation.
If a structural feature is functionally important, it is typically well conserved in groups of related RNAs, even if the level of primary sequence conservation may be low.One sequence can have multiple RNA secondary structures, transient structures
Sunday, September 26, 2010
The Nuclear-Retained Noncoding RNA MALAT1 Regulates Alternative Splicing by Modulating SR Splicing Factor Phosphorylation
http://www.sciencedaily.com/releases/2010/09/100923162412.htm?utm_source=feedburner&utm_medium=feed&utm_campaign=Feed%3A+sciencedaily+(ScienceDaily%3A+Latest+Science+News)
http://www.cell.com/molecular-cell/retrieve/pii/S1097276510006210
Researchers report this month that MALAT1, a long non-coding RNA that is implicated in certain cancers, regulates pre-mRNA splicing -- a critical step in the earliest stage of protein production. Their study appears in the journal Molecular Cell.
http://www.cell.com/molecular-cell/retrieve/pii/S1097276510006210
Researchers report this month that MALAT1, a long non-coding RNA that is implicated in certain cancers, regulates pre-mRNA splicing -- a critical step in the earliest stage of protein production. Their study appears in the journal Molecular Cell.
Stack vs Heap memory space
http://en.wikipedia.org/wiki/Stack-based_memory_allocation
In most modern computer systems, each thread has a reserved region of memory referred to as its stack. When a function executes, it may add some of its state data to the top of the stack; when the function exits it is responsible for removing that data from the stack. At a minimum, a thread's stack is used to store the location of function calls in order to allow return statements to return to the correct location, but programmers may further choose to explicitly use the stack. If a region of memory lies on the thread's stack, that memory is said to have been allocated on the stack.
Because the data is added and removed in a last-in-first-out manner, stack allocation is very simple and typically faster than heap-based memory allocation (also known as dynamic memory allocation). Another feature is that memory on the stack is automatically, and very efficiently, reclaimed when the function exits, which can be convenient for the programmer if the data is no longer required. If however, the data needs to be kept in some form, then it must be copied from the stack before the function exits. Therefore, stack based allocation is suitable for temporary data or data which is no longer required after the creating function exits.
A disadvantage of stack-based memory allocation is that a thread's stack size can be as small as a few dozen kilobytes. Allocating more memory on the stack than is available can result in a crash due to stack overflow.
In most modern computer systems, each thread has a reserved region of memory referred to as its stack. When a function executes, it may add some of its state data to the top of the stack; when the function exits it is responsible for removing that data from the stack. At a minimum, a thread's stack is used to store the location of function calls in order to allow return statements to return to the correct location, but programmers may further choose to explicitly use the stack. If a region of memory lies on the thread's stack, that memory is said to have been allocated on the stack.
Because the data is added and removed in a last-in-first-out manner, stack allocation is very simple and typically faster than heap-based memory allocation (also known as dynamic memory allocation). Another feature is that memory on the stack is automatically, and very efficiently, reclaimed when the function exits, which can be convenient for the programmer if the data is no longer required. If however, the data needs to be kept in some form, then it must be copied from the stack before the function exits. Therefore, stack based allocation is suitable for temporary data or data which is no longer required after the creating function exits.
A disadvantage of stack-based memory allocation is that a thread's stack size can be as small as a few dozen kilobytes. Allocating more memory on the stack than is available can result in a crash due to stack overflow.
microRNAs: Master Regulators as Potential Therapeutics in Cancer
microRNAs: Master Regulators as Potential Therapeutics in Cancer
Michela Garofalo and Carlo M. Croce
Department of Molecular Virology, Immunology and Medical Genetics and Comprehensive
Cancer Center, Ohio State University, Columbus, Ohio 43210;
email: michela.garofalo@osumc.edu, carlo.croce@osumc.edu
http://www.annualreviews.org/doi/abs/10.1146/annurev-pharmtox-010510-100517
Cell. 2009 Jan 23;136(2):215-33.
MicroRNAs: target recognition and regulatory functions.
Bartel DP.
http://www.ncbi.nlm.nih.gov/pubmed/19167326
http://download.cell.com/pdf/PIIS0092867409000087.pdf
Many miRNAs have been shown to function as oncogenes in the majority of cancers profiled to date (Table 1). miR-155 was one of the first described (30).
miR-155, miR-21, miR-221&222, miR-106b-93-25 cluster, miR-17-92 cluster
The tumor suppressor function of miR-15a/16-1 has also been addressed
in vivo.
The most promising therapeutic techniques tested to date are
(a) miRNA mimics and (b) anti-miRNA oligonucleotides (AMOs).
http://www.youtube.com/watch?v=oANi7PRqalM
TAM: A method for enrichment and depletion analysis of a microRNA category in a list of microRNAs
Ming Lu1 Bing Shi1,2 Juan Wang1 Qun Cao1 Qinghua Cui1
http://www.biomedcentral.com/1471-2105/11/419
TargetSpy: a supervised machine learning approach for microRNA target prediction
Martin Sturm1 Michael Hackenberg2 David Langenberger3,4 Dmitrij Frishman1,4
http://www.biomedcentral.com/1471-2105/11/292
Michela Garofalo and Carlo M. Croce
Department of Molecular Virology, Immunology and Medical Genetics and Comprehensive
Cancer Center, Ohio State University, Columbus, Ohio 43210;
email: michela.garofalo@osumc.edu, carlo.croce@osumc.edu
http://www.annualreviews.org/doi/abs/10.1146/annurev-pharmtox-010510-100517
Cell. 2009 Jan 23;136(2):215-33.
MicroRNAs: target recognition and regulatory functions.
Bartel DP.
http://www.ncbi.nlm.nih.gov/pubmed/19167326
http://download.cell.com/pdf/PIIS0092867409000087.pdf
Many miRNAs have been shown to function as oncogenes in the majority of cancers profiled to date (Table 1). miR-155 was one of the first described (30).
miR-155, miR-21, miR-221&222, miR-106b-93-25 cluster, miR-17-92 cluster
The tumor suppressor function of miR-15a/16-1 has also been addressed
in vivo.
The most promising therapeutic techniques tested to date are
(a) miRNA mimics and (b) anti-miRNA oligonucleotides (AMOs).
http://www.youtube.com/watch?v=oANi7PRqalM
TAM: A method for enrichment and depletion analysis of a microRNA category in a list of microRNAs
Ming Lu1 Bing Shi1,2 Juan Wang1 Qun Cao1 Qinghua Cui1
http://www.biomedcentral.com/1471-2105/11/419
TargetSpy: a supervised machine learning approach for microRNA target prediction
Martin Sturm1 Michael Hackenberg2 David Langenberger3,4 Dmitrij Frishman1,4
http://www.biomedcentral.com/1471-2105/11/292
Genomewide Association Studies and Assessment of the Risk of Disease
http://www.nejm.org/doi/full/10.1056/NEJMra0905980
Nearly 600 genomewide association studies covering 150 distinct diseases and traits have been published, with nearly 800 SNP–trait associations reported as significant (P<5×10−8)
Approximately 40% of trait-associated SNPs fall in intergenic regions, and another 40% are located in noncoding introns. These two findings have sharpened the focus on the potential roles of intronic, and particularly intergenic, regions in regulating gene expression. 1
Although intronic and intergenic SNPs are not overrepresented in associations as compared with randomly selected SNPs, they account for the great majority — more than 80% — of associated SNPs.
Given the lack of good representation of SNPs with a prevalence of less than 5% in current genomewide association arrays, a comprehensive catalogue of SNPs with a prevalence of 1 to 5% is being generated by the 1000 Genomes Project55 for potential inclusion in fine-mapping efforts and expanded genomewide association arrays. In the project's pilot effort, more than 11 million novel SNPs have been identified in what was initially low-depth coverage of 172 persons.
Annotation catalogues (maps of functions of variants), such as those related to transcription-factor binding (promoting gene expression) or to RNA interference (silencing genes), are currently in development and should facilitate the identification of functional variants underlying genomewide association signals.57
The importance of structural variation, including copy-number variants, inversions, and translocations, is an active area of investigation; several structural variants underlie genomewide association signals for autism, schizophrenia, Crohn's disease, and obesity.
For the prediction of complex diseases, genotypes at multiple SNPs are often combined into scores calculated according to the number of risk alleles carried, which is the approach that Kathiresan and colleagues used in predicting the risk of cardiovascular disease on the basis of nine SNPs associated with cholesterol levels
What is becoming clear from these early attempts at genetically based risk assessment is that currently known variants explain too little about the risk of disease occurrence to be of clinically useful predictive value.
Possible clinical uses of predictive scores — for example, in deciding which patients should be screened more intensively for breast cancer with the use of mammography69 or for statin-induced myopathy with the use of muscle enzyme assays70 — will require rigorous, preferably prospective, evaluation before being accepted into clinical practice.
The ability to assess risk for 120 conditions at the same time also raises the concern that predictive models will yield conflicting recommendations; if implemented, they could reduce a person's risk for development of one condition and exacerbate the risk for development of another.
Patients inquiring about genomewide association testing should be advised that at present the results of such testing have no value in predicting risk and are not clinically directive.
Much more remains to be learned about how variations in intronic and intergenic regions (where the vast majority of SNP–trait associations reside) influence gene expression, protein coding, and disease phenotypes.
The substantial challenges of incorporating such research into clinical care must be pursued if the potential of genomic medicine is to be realized.
Hardy J, Singleton A. Genomewide association studies and human disease. N Engl J Med 2009;360:1759-1768 Full Text | Web of Science | Medline http://www.ncbi.nlm.nih.gov/pubmed/19369657
http://www.sfu.ca/~chenn/2010.html
Nearly 600 genomewide association studies covering 150 distinct diseases and traits have been published, with nearly 800 SNP–trait associations reported as significant (P<5×10−8)
Approximately 40% of trait-associated SNPs fall in intergenic regions, and another 40% are located in noncoding introns. These two findings have sharpened the focus on the potential roles of intronic, and particularly intergenic, regions in regulating gene expression. 1
Although intronic and intergenic SNPs are not overrepresented in associations as compared with randomly selected SNPs, they account for the great majority — more than 80% — of associated SNPs.
Given the lack of good representation of SNPs with a prevalence of less than 5% in current genomewide association arrays, a comprehensive catalogue of SNPs with a prevalence of 1 to 5% is being generated by the 1000 Genomes Project55 for potential inclusion in fine-mapping efforts and expanded genomewide association arrays. In the project's pilot effort, more than 11 million novel SNPs have been identified in what was initially low-depth coverage of 172 persons.
Annotation catalogues (maps of functions of variants), such as those related to transcription-factor binding (promoting gene expression) or to RNA interference (silencing genes), are currently in development and should facilitate the identification of functional variants underlying genomewide association signals.57
The importance of structural variation, including copy-number variants, inversions, and translocations, is an active area of investigation; several structural variants underlie genomewide association signals for autism, schizophrenia, Crohn's disease, and obesity.
For the prediction of complex diseases, genotypes at multiple SNPs are often combined into scores calculated according to the number of risk alleles carried, which is the approach that Kathiresan and colleagues used in predicting the risk of cardiovascular disease on the basis of nine SNPs associated with cholesterol levels
What is becoming clear from these early attempts at genetically based risk assessment is that currently known variants explain too little about the risk of disease occurrence to be of clinically useful predictive value.
Possible clinical uses of predictive scores — for example, in deciding which patients should be screened more intensively for breast cancer with the use of mammography69 or for statin-induced myopathy with the use of muscle enzyme assays70 — will require rigorous, preferably prospective, evaluation before being accepted into clinical practice.
The ability to assess risk for 120 conditions at the same time also raises the concern that predictive models will yield conflicting recommendations; if implemented, they could reduce a person's risk for development of one condition and exacerbate the risk for development of another.
Patients inquiring about genomewide association testing should be advised that at present the results of such testing have no value in predicting risk and are not clinically directive.
Much more remains to be learned about how variations in intronic and intergenic regions (where the vast majority of SNP–trait associations reside) influence gene expression, protein coding, and disease phenotypes.
The substantial challenges of incorporating such research into clinical care must be pursued if the potential of genomic medicine is to be realized.
Hardy J, Singleton A. Genomewide association studies and human disease. N Engl J Med 2009;360:1759-1768 Full Text | Web of Science | Medline http://www.ncbi.nlm.nih.gov/pubmed/19369657
http://www.sfu.ca/~chenn/2010.html
6 comments
Glad to know that at least 6 ppl had read my posts ^_^.
In the blog, go to Design and there's the comments tab. Another cool thing is the Stats tab. I was pretty amazed that most users use IE on Windows from Russia! Welcome! приветствовать
In the blog, go to Design and there's the comments tab. Another cool thing is the Stats tab. I was pretty amazed that most users use IE on Windows from Russia! Welcome! приветствовать
Logarithmic transformation
http://www.statsdirect.com/help/data_preparation/log_trans.htm
Effects of log transformation:
* Variance stabilisation.
* Increasing slopes in x in relation to another variable are linearized.
* Positively skewed distributions of x are normalized.
The function named Log (natural) calculates the natural (Naperian, log to the base e) logarithm of the data you select.
The function named Log (base 10) calculates the common (log to the base 10) logarithm of the data you select.
Effects of log transformation:
* Variance stabilisation.
* Increasing slopes in x in relation to another variable are linearized.
* Positively skewed distributions of x are normalized.
The function named Log (natural) calculates the natural (Naperian, log to the base e) logarithm of the data you select.
The function named Log (base 10) calculates the common (log to the base 10) logarithm of the data you select.
Saturday, September 25, 2010
Eye bags
http://www.getridofthings.com/get-rid-of-bags-under-eyes.htm
http://facialexercisesguide.com/19/eye-bags-how-to-get-rid-of-bags-under-eyes/
cause: genetics, water retention under the eyes due to dehydration or being tired
Using a cold compress for 10 - 15 minutes, one to two times daily, will help get rid of the bags under your eyes for a short period
Out of all the bizarre folk remedies, application of frozen or chilled green tea bags may be one of the few that can actually get rid of bags under eyes.
You may need to address your water retention to get rid of the bags under your eyes.
http://facialexercisesguide.com/19/eye-bags-how-to-get-rid-of-bags-under-eyes/
cause: genetics, water retention under the eyes due to dehydration or being tired
Using a cold compress for 10 - 15 minutes, one to two times daily, will help get rid of the bags under your eyes for a short period
Out of all the bizarre folk remedies, application of frozen or chilled green tea bags may be one of the few that can actually get rid of bags under eyes.
You may need to address your water retention to get rid of the bags under your eyes.
Friday, September 24, 2010
Rails Cheat Sheet
Rails file structure notes
* Gemfile - which libraries are to be installed and used, the ‘gem’ collections
group :development do
gem "rspec-rails", ">= 2.0.0.beta.22"
end
* config/routes.rb - when a new view has been added, named routes
match '/help', :to => 'pages#help' # about_path => '/about', about_url => 'http://localhost:3000/about'
o root :to => 'pages#home' # This code maps the root URL / to /pages/home
* app/controllers/pages_controller.rb - when a new page has been added, Ruby code here
def help
@title = "Help"
end
* app/views/pages/help.html.erb - view
* app/views/layouts/application.html.erb - the master html where to reference header and footer partials
<%= render 'layouts/header' %> # reference _header.html.erb partial
* public/stylesheets/custom.css - public stylesheets
nav ul li {
list-style-type: disc;
display: inline-block;
padding: 0.2em 0;
}
* public/images/logo.png - where images are stored
* app/views/layouts/_header.html.erb - partials, ie header, footer, CSS
<%= link_to 'Help', help_path %>
# use help_path named route
<%= stylesheet_link_tag 'custom', :media => 'screen' %> # use custom.css
* $ rails generate controller Users new - create a Users controller with action new
create app/controllers/users_controller.rb
route get "users/new"
* $ rails generate model User name:string email:string - generate model, same as above, model names are singular and controller names are plural
* spec/controllers/users_controller_spec.rb - tests Users controller
it "should be successful" do
get '/signup' response.should be_success
response.should have_selector('title', :content => "Sign up")
end
* $ rspec spec/ - run tests
* db/migrate/_create_users.rb - User model (database)
o class CreateUsers < ActiveRecord::Migration def self.up create_table :users do |t| t.string :name
* $ rake db:migrate - migrate up, applies changes in db/*/*.rb model files and calls self.up to create a file called db/development.sqlite3
* $ rake db:rollback - tear down, ie. calls self.down
* $ rails console --sandbox - loads console with read-only access to DB
>> foo = User.create(:name => "Foo", :email => "foo@bar.com")
=> #
>> user.errors.full_messages - display error message for object user in rails console
=> ["Name can't be blank"]
* $ tail -f log/development.log - console DB logs
* app/models/user.rb - Model attribute validate
validates :name, :presence => true
* $ rails generate migration add_email_uniqueness_index - adding structure to an existing model, which creates db/migrate/_add_email_uniqueness_index.rb
* Gemfile - which libraries are to be installed and used, the ‘gem’ collections
group :development do
gem "rspec-rails", ">= 2.0.0.beta.22"
end
* config/routes.rb - when a new view has been added, named routes
match '/help', :to => 'pages#help' # about_path => '/about', about_url => 'http://localhost:3000/about'
o root :to => 'pages#home' # This code maps the root URL / to /pages/home
* app/controllers/pages_controller.rb - when a new page has been added, Ruby code here
def help
@title = "Help"
end
* app/views/pages/help.html.erb - view
* app/views/layouts/application.html.erb - the master html where to reference header and footer partials
<%= render 'layouts/header' %> # reference _header.html.erb partial
* public/stylesheets/custom.css - public stylesheets
nav ul li {
list-style-type: disc;
display: inline-block;
padding: 0.2em 0;
}
* public/images/logo.png - where images are stored
* app/views/layouts/_header.html.erb - partials, ie header, footer, CSS
<%= link_to 'Help', help_path %>
# use help_path named route
<%= stylesheet_link_tag 'custom', :media => 'screen' %> # use custom.css
* $ rails generate controller Users new - create a Users controller with action new
create app/controllers/users_controller.rb
route get "users/new"
* $ rails generate model User name:string email:string - generate model, same as above, model names are singular and controller names are plural
* spec/controllers/users_controller_spec.rb - tests Users controller
it "should be successful" do
get '/signup' response.should be_success
response.should have_selector('title', :content => "Sign up")
end
* $ rspec spec/ - run tests
* db/migrate/
o class CreateUsers < ActiveRecord::Migration def self.up create_table :users do |t| t.string :name
=> #
>> user.errors.full_messages - display error message for object user in rails console
=> ["Name can't be blank"]
validates :name, :presence => true
lorem ipsum - filler text for web pages
http://www.straightdope.com/columns/read/2290/what-does-the-filler-text-lorem-ipsum-mean
"Turns out the passage doesn't just look like real Latin, it is real (although slightly scrambled), and from a famous source. This news came from Richard McClintock, a Latin professor turned publications director at Hampden-Sydney College in Virginia. Curious about what the words meant, McClintock had looked up one of the more obscure ones, consectetur, in a Latin dictionary. Going through the cites of the word in classical literature, he found one that looked familiar. Aha! Lorem ipsum was part of a passage from Cicero, specifically De finibus bonorum et malorum, a treatise on the theory of ethics written in 45 BC. The original reads, Neque porro quisquam est qui dolorem ipsum quia dolor sit amet, consectetur, adipisci velit . . . ("There is no one who loves pain itself, who seeks after it and wants to have it, simply because it is pain . . .").
McClintock recalled having seen lorem ipsum in a book of early metal type samples, which commonly used extracts from the classics. "What I find remarkable," he told B&A, "is that this text has been the industry's standard dummy text ever since some printer in the 1500s took a galley of type and scrambled it to make a type specimen book; it has survived not only four centuries of letter-by-letter resetting but even the leap into electronic typesetting, essentially unchanged." So much for the transitory nature of content in the information age. "
... But it's easier to believe that someone at Letraset simply copied the text from an old hot-type source.
- Cecil Adams
"Turns out the passage doesn't just look like real Latin, it is real (although slightly scrambled), and from a famous source. This news came from Richard McClintock, a Latin professor turned publications director at Hampden-Sydney College in Virginia. Curious about what the words meant, McClintock had looked up one of the more obscure ones, consectetur, in a Latin dictionary. Going through the cites of the word in classical literature, he found one that looked familiar. Aha! Lorem ipsum was part of a passage from Cicero, specifically De finibus bonorum et malorum, a treatise on the theory of ethics written in 45 BC. The original reads, Neque porro quisquam est qui dolorem ipsum quia dolor sit amet, consectetur, adipisci velit . . . ("There is no one who loves pain itself, who seeks after it and wants to have it, simply because it is pain . . .").
McClintock recalled having seen lorem ipsum in a book of early metal type samples, which commonly used extracts from the classics. "What I find remarkable," he told B&A, "is that this text has been the industry's standard dummy text ever since some printer in the 1500s took a galley of type and scrambled it to make a type specimen book; it has survived not only four centuries of letter-by-letter resetting but even the leap into electronic typesetting, essentially unchanged." So much for the transitory nature of content in the information age. "
... But it's easier to believe that someone at Letraset simply copied the text from an old hot-type source.
- Cecil Adams
Artificial Neural Network
http://en.wikipedia.org/wiki/Artificial_neural_network
An artificial neural network (ANN), usually called "neural network" (NN), is a mathematical model or computational model that is inspired by the structure and/or functional aspects of biological neural networks. It consists of an interconnected group of artificial neurons and processes information using a connectionist approach to computation. In mosts cases an ANN is an adaptive system that changes its structure based on external or internal information that flows through the network during the learning phase. Modern neural networks are non-linear statistical data modeling tools. They are usually used to model complex relationships between inputs and outputs or to find patterns in data.
http://www.faqs.org/faqs/ai-faq/neural-nets/part1/section-11.html
SOM
http://en.wikipedia.org/wiki/Self-organizing_map
A self-organizing map (SOM) or self-organizing feature map (SOFM) is a type of artificial neural network that is trained using unsupervised learning to produce a low-dimensional (typically two-dimensional), discretized representation of the input space of the training samples, called a map. Self-organizing maps are different from other artificial neural networks in the sense that they use a neighborhood function to preserve the topological properties of the input space.
LVQ
LVQ can be understood as a special case of an artificial neural network, more precisely, it applies a winner-take-all Hebbian learning-based approach. It is a precursor to Self-organizing maps (SOM) and related to Neural gas, and to the k-Nearest Neighbor algorithm (k-NN). LVQ was invented by Teuvo Kohonen.
An advantage of LVQ is that it creates prototypes that are easy to interpret for experts in the field.[citation needed]
An artificial neural network (ANN), usually called "neural network" (NN), is a mathematical model or computational model that is inspired by the structure and/or functional aspects of biological neural networks. It consists of an interconnected group of artificial neurons and processes information using a connectionist approach to computation. In mosts cases an ANN is an adaptive system that changes its structure based on external or internal information that flows through the network during the learning phase. Modern neural networks are non-linear statistical data modeling tools. They are usually used to model complex relationships between inputs and outputs or to find patterns in data.
http://www.faqs.org/faqs/ai-faq/neural-nets/part1/section-11.html
SOM
http://en.wikipedia.org/wiki/Self-organizing_map
A self-organizing map (SOM) or self-organizing feature map (SOFM) is a type of artificial neural network that is trained using unsupervised learning to produce a low-dimensional (typically two-dimensional), discretized representation of the input space of the training samples, called a map. Self-organizing maps are different from other artificial neural networks in the sense that they use a neighborhood function to preserve the topological properties of the input space.
LVQ
LVQ can be understood as a special case of an artificial neural network, more precisely, it applies a winner-take-all Hebbian learning-based approach. It is a precursor to Self-organizing maps (SOM) and related to Neural gas, and to the k-Nearest Neighbor algorithm (k-NN). LVQ was invented by Teuvo Kohonen.
An advantage of LVQ is that it creates prototypes that are easy to interpret for experts in the field.[citation needed]
Linear models - STAT 545
http://www.stat.ubc.ca/~gustaf/stat545
Paul Gustafson
STATISTICS 545 (2005-2006, Term 1)
Course Outline (REVISED VERSION OF SEPT. 6, pdf file)
Assigned coursework (will be added onto as we cover material).
Page last updated: Nov. 22, 2005
Lecture 1: Statistical Principles I: Estimation (handwritten notes).
Lecture 2: Statistical Principles II: Uncertainty assessment and hypothesis testing (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 3: Nice things about R (slides - pdf: fullsize, reduced, ps: fullsize, reduced ), and three example tasks (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 4: Linear models, Part I (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 5: Linear models, Part II: handwritten slides on regression diagnostics.
Lecture 6: Logistic regression (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 7: Generalized linear models I (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 8: Generalized linear models II - Overdispersion (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 9: Generalized linear models III - Log-linear modelling (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
We lapped ourselves. It took us 10 classes to cover lectures 1 through 9. Hence there is no lecture 10.
Lecture 11: The bootstrap (slides - pdf: fullsize, reduced, ps: fullsize, reduced ). Also, here is R code for examples one, two, and three.
Lecture 12: Model Choice (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 13: More Model Choice (slides - pdf: fullsize, reduced, ps: fullsize, reduced ). Also, R code for the stepwise and cross-validation examples.
Lapped ourselves again - 4 classes to cover lectures 11 through 13 - Hence there is no lecture 14.
Lecture 15: Nonlinear Regression (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 16: Expectation-Maximization (EM) Algorithm (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 17: Simulation Studies (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 18: Handwritten notes on hierarchical models (otherwise known as random-effect models, mixed models, or random coefficient models).
Lecture 19: More on hierarchical models, including the Sitka data ex.: (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 20: Curve-fitting. No pre-fab slides, but some pictures ( pdf, or ps), and a bit of code.
Lecture 21: Move from curve-fitting to additive models, with this example: (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 22: We didn't really get started on additive models last time, so that discussion carrys over. We may also start talking about "tree models" if time permits.
Lecture 23: Tree models. Here are the examples. ( pdf: fullsize, reduced, ps: fullsize, reduced ).
Paul Gustafson
STATISTICS 545 (2005-2006, Term 1)
Course Outline (REVISED VERSION OF SEPT. 6, pdf file)
Assigned coursework (will be added onto as we cover material).
Page last updated: Nov. 22, 2005
Lecture 1: Statistical Principles I: Estimation (handwritten notes).
Lecture 2: Statistical Principles II: Uncertainty assessment and hypothesis testing (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 3: Nice things about R (slides - pdf: fullsize, reduced, ps: fullsize, reduced ), and three example tasks (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 4: Linear models, Part I (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 5: Linear models, Part II: handwritten slides on regression diagnostics.
Lecture 6: Logistic regression (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 7: Generalized linear models I (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 8: Generalized linear models II - Overdispersion (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 9: Generalized linear models III - Log-linear modelling (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
We lapped ourselves. It took us 10 classes to cover lectures 1 through 9. Hence there is no lecture 10.
Lecture 11: The bootstrap (slides - pdf: fullsize, reduced, ps: fullsize, reduced ). Also, here is R code for examples one, two, and three.
Lecture 12: Model Choice (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 13: More Model Choice (slides - pdf: fullsize, reduced, ps: fullsize, reduced ). Also, R code for the stepwise and cross-validation examples.
Lapped ourselves again - 4 classes to cover lectures 11 through 13 - Hence there is no lecture 14.
Lecture 15: Nonlinear Regression (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 16: Expectation-Maximization (EM) Algorithm (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 17: Simulation Studies (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 18: Handwritten notes on hierarchical models (otherwise known as random-effect models, mixed models, or random coefficient models).
Lecture 19: More on hierarchical models, including the Sitka data ex.: (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 20: Curve-fitting. No pre-fab slides, but some pictures ( pdf, or ps), and a bit of code.
Lecture 21: Move from curve-fitting to additive models, with this example: (slides - pdf: fullsize, reduced, ps: fullsize, reduced ).
Lecture 22: We didn't really get started on additive models last time, so that discussion carrys over. We may also start talking about "tree models" if time permits.
Lecture 23: Tree models. Here are the examples. ( pdf: fullsize, reduced, ps: fullsize, reduced ).
penultimate
adj.
1. Next to last.
2. Linguistics. Of or relating to the penult of a word: penultimate stress.
n.
The next to the last.
1. Next to last.
2. Linguistics. Of or relating to the penult of a word: penultimate stress.
n.
The next to the last.
Interlogs - interaction is conserved between orthologs
conserved protein-protein interactions
http://www.biomedcentral.com/pubmed/11731503
http://ymbc.ym.edu.tw/proteome/interact/interlog.htm
http://www.biomedcentral.com/pubmed/11731503
http://ymbc.ym.edu.tw/proteome/interact/interlog.htm
Genetic markers
http://en.wikipedia.org/wiki/Genetic_marker
A genetic marker is a gene or DNA sequence with a known location on a chromosome that can be used to identify cells, individuals or species. It can be described as a variation (which may arise due to mutation or alteration in the genomic loci) that can be observed. A genetic marker may be a short DNA sequence, such as a sequence surrounding a single base-pair change (single nucleotide polymorphism, SNP), or a long one, like minisatellites. (short tandem repeat)
A genetic marker is a gene or DNA sequence with a known location on a chromosome that can be used to identify cells, individuals or species. It can be described as a variation (which may arise due to mutation or alteration in the genomic loci) that can be observed. A genetic marker may be a short DNA sequence, such as a sequence surrounding a single base-pair change (single nucleotide polymorphism, SNP), or a long one, like minisatellites. (short tandem repeat)
Wednesday, September 22, 2010
MEGAN - Metagenome Analysis Software
MEGAN - http://www-ab.informatik.uni-tuebingen.de/software/megan
primer walk, shotgun
Solexa sequencing
http://awcgs.massey.ac.nz/seqserver/solexa-faq.htm
Dideoxy chain-termination sequencing depends on synthetic DNA primer sequences to initiate the reaction. These primers must match a portion of the template whose sequence we are trying to determine. This gives us a 'chicken and egg' problem of needing to know a bit of the template sequence before we can read more of it.
One way to start sequencing an unknown sequence is to make a recombinant clone, putting the unknown insert into a vector of known sequence. Then primers from the vector can be used to begin reading the sequence of the insert. Once a portion of the new insert sequence is known, we can use that to design a new primer to let us read further. This process can be repeated until the whole insert is sequenced. This 'primer walking' process is inherently sequential, since each step must be completed before the results can be used to design the primer for the next step.
Shotgun sequencing is an approach that lets us run large numbers of reactions in parallel, rather than in series. Rather than using primer walking through one large insert, we randomly fragment the insert to create a library of smaller fragments. A large number of these clones are chosen at random, and sequenced in parallel using primers matching the vector. The sequencing results are then 'assembled' on the computer into a contiguous sequence of overlapping fragments. This approach essentially trades much of the laborious laboratory work for a puzzle to be solved on the computer, and turns out to be much faster than pure primer walking.
http://awcgs.massey.ac.nz/seqserver/solexa-faq.htm
Dideoxy chain-termination sequencing depends on synthetic DNA primer sequences to initiate the reaction. These primers must match a portion of the template whose sequence we are trying to determine. This gives us a 'chicken and egg' problem of needing to know a bit of the template sequence before we can read more of it.
One way to start sequencing an unknown sequence is to make a recombinant clone, putting the unknown insert into a vector of known sequence. Then primers from the vector can be used to begin reading the sequence of the insert. Once a portion of the new insert sequence is known, we can use that to design a new primer to let us read further. This process can be repeated until the whole insert is sequenced. This 'primer walking' process is inherently sequential, since each step must be completed before the results can be used to design the primer for the next step.
Shotgun sequencing is an approach that lets us run large numbers of reactions in parallel, rather than in series. Rather than using primer walking through one large insert, we randomly fragment the insert to create a library of smaller fragments. A large number of these clones are chosen at random, and sequenced in parallel using primers matching the vector. The sequencing results are then 'assembled' on the computer into a contiguous sequence of overlapping fragments. This approach essentially trades much of the laborious laboratory work for a puzzle to be solved on the computer, and turns out to be much faster than pure primer walking.
Tuesday, September 21, 2010
Windows can't ping
I had some issues pining a wireless printer, funny thing is I've tried a couple of things, restarts, nothing worked. The other computer connected to the network can ping it.
Finally after I powered down the laptop and turn it back on, it finally worked! So it's either you need to power down the computer (not just restarting) or you just have to wait for say 10 - 20 min.?
Finally after I powered down the laptop and turn it back on, it finally worked! So it's either you need to power down the computer (not just restarting) or you just have to wait for say 10 - 20 min.?
Ruby
http://railstutorial.org/chapters/rails-flavored-ruby#top
Ruby loop
$ rails console --sandbox
> a = %w[12@bc.com the_user@bcc.ca bla@blah.com]
> a.each do |a|
> puts a
> end
12@bc.com
the_user@bcc.ca
bla@blah.com
=> ["12@bc.com", "the_user@bcc.ca", "bla@blah.com"]
blocks
>> (1..5).each { |i| puts 2 * i }
2
4
6
8
10
=> 1..5
>> (1..5).each do |i|
?> puts 2 * i
>> end
2
4
6
8
10
=> 1..5
>> ret = (1..5).map { |i| i**2 } # The ** notation is for 'power'., stores value to 'ret'
=> [1, 4, 9, 16, 25]
so basically, symbols, eg. :name and instances @name does not throw exceptions
You can modify classes:
>> class String
>> # Return true if the string is its own reverse.
>> def palindrome?
>> self == self.reverse
>> end
>> end
=> nil
>> "deified".palindrome?http://railstutorial.org/chapters/filling-in-the-layout#top
=> true
Setting up rspec and sqlite3 can be a pain if you're using wrong versions while trying to follow tutorials ...
Ruby loop
$ rails console --sandbox
> a = %w[12@bc.com the_user@bcc.ca bla@blah.com]
> a.each do |a|
> puts a
> end
12@bc.com
the_user@bcc.ca
bla@blah.com
=> ["12@bc.com", "the_user@bcc.ca", "bla@blah.com"]
blocks
>> (1..5).each { |i| puts 2 * i }
2
4
6
8
10
=> 1..5
>> (1..5).each do |i|
?> puts 2 * i
>> end
2
4
6
8
10
=> 1..5
>> ret = (1..5).map { |i| i**2 } # The ** notation is for 'power'., stores value to 'ret'
=> [1, 4, 9, 16, 25]
so basically, symbols, eg. :name and instances @name does not throw exceptions
You can modify classes:
>> class String
>> # Return true if the string is its own reverse.
>> def palindrome?
>> self == self.reverse
>> end
>> end
=> nil
>> "deified".palindrome?http://railstutorial.org/chapters/filling-in-the-layout#top
=> true
Setting up rspec and sqlite3 can be a pain if you're using wrong versions while trying to follow tutorials ...
The Unreasonable Effectiveness of Google
http://duncan.hull.name/2009/04/17/the-unreasonable-effectiveness-of-google/
http://googleresearch.blogspot.com/2009/03/unreasonable-effectiveness-of-data.html
http://www.biomodels.net/
http://www.sbml.org/
The paper concludes:
“So, follow the data. Choose a representation that can use unsupervised learning on unlabeled data, which is so much more plentiful than labeled data. Represent all the data with a nonparametric model rather than trying to summarize it with a parametric model, because with very large data sources, the data holds a lot of detail. For natural language applications, trust that human language has already evolved words for the important concepts. See how far you can go by tying together the words that are already there, rather than by inventing new concepts with clusters of words. Now go out and gather some data, and see what it can do.”
http://googleresearch.blogspot.com/2009/03/unreasonable-effectiveness-of-data.html
http://www.biomodels.net/
http://www.sbml.org/
The paper concludes:
“So, follow the data. Choose a representation that can use unsupervised learning on unlabeled data, which is so much more plentiful than labeled data. Represent all the data with a nonparametric model rather than trying to summarize it with a parametric model, because with very large data sources, the data holds a lot of detail. For natural language applications, trust that human language has already evolved words for the important concepts. See how far you can go by tying together the words that are already there, rather than by inventing new concepts with clusters of words. Now go out and gather some data, and see what it can do.”
Rspec: Command not found
> rvm use 1.9.2@rails3tutorial --default
The sanity check would then be to open up a new terminal window and check which ruby you're using along with your gem directory:
> which ruby
--> /Users/doug/.rvm/rubies/ruby-1.9.2-rc2/bin/ruby
> rvm gemdir
--> /Users/doug/.rvm/gems/ruby-1.9.2-rc2@rails3tutorial
If this is all working, then when you run "gem install blah" (without sudo!) then the blah gem will be installed in the rails3tutorial gem directory.
put this at the top of ~/.bashrc
[[ -s "$HOME/.rvm/scripts/rvm" ]] && source "$HOME/.rvm/scripts/rvm" # This loads RVM into a shell session.
$ rvm info
$ rvm install 1.9.2
$ rvm update
$ rvm use 1.9.2
$ rvm list known
http://docs.rubygems.org/read/chapter/10
seems that ruby 1.9.2 is a bit buggy (sqlite3 segmentation fault in ruby 1.9.2), use 1.8.7 for now
The sanity check would then be to open up a new terminal window and check which ruby you're using along with your gem directory:
> which ruby
--> /Users/doug/.rvm/rubies/ruby-1.9.2-rc2/bin/ruby
> rvm gemdir
--> /Users/doug/.rvm/gems/ruby-1.9.2-rc2@rails3tutorial
If this is all working, then when you run "gem install blah" (without sudo!) then the blah gem will be installed in the rails3tutorial gem directory.
put this at the top of ~/.bashrc
[[ -s "$HOME/.rvm/scripts/rvm" ]] && source "$HOME/.rvm/scripts/rvm" # This loads RVM into a shell session.
$ rvm info
$ rvm install 1.9.2
$ rvm update
$ rvm use 1.9.2
$ rvm list known
http://docs.rubygems.org/read/chapter/10
seems that ruby 1.9.2 is a bit buggy (sqlite3 segmentation fault in ruby 1.9.2), use 1.8.7 for now
curl to issue HTTP requests
If you install curl, a command-line client that can issue HTTP requests, you can see this directly at, e.g., www.google.com (where the --head flag prevents curl from returning the whole page):
$ curl --head www.google.com
HTTP/1.1 200 OK
.
.
$ curl --head www.google.com
HTTP/1.1 302 Found
Location: http://www.google.ca/
Cache-Control: private
Content-Type: text/html; charset=UTF-8
$ curl www.google.com
HTTP/1.1 302 Found
Location: http://www.google.ca/
Cache-Control: private
Content-Type: text/html; charset=UTF-8
$ curl www.google.com
The document has moved
here.
$ curl --head www.google.com
HTTP/1.1 200 OK
.
.
$ curl --head www.google.com
HTTP/1.1 302 Found
Location: http://www.google.ca/
Cache-Control: private
Content-Type: text/html; charset=UTF-8
$ curl www.google.com
HTTP/1.1 302 Found
Location: http://www.google.ca/
Cache-Control: private
Content-Type: text/html; charset=UTF-8
$ curl www.google.com
302 Moved
The document has moved
here.
Rails 3.0 Changes
Rails 2.2 vs Rails 3.0 Changes
http://www.pragprog.com/wikis/wiki/ChangesInRails30
Global changes
- The names of scripts have changed
http://www.pragprog.com/wikis/wiki/ChangesInRails30
Global changes
- The names of scripts have changed
Rails 2.2 ----> Rails 3.0
rails ----> rails new
ruby script/about ----> rake about
ruby script/console ----> rails console
ruby script/dbconsole ----> rails dbconsole
ruby script/destroy ----> rails destroy
ruby script/performance/benchmarker ----> rails benchmarker
ruby script/performance/profiler ----> rails profiler
ruby script/generate ----> rails generate
ruby script/plugin ----> rails plugin
ruby script/runner ----> rails runner
ruby script/server ----> rails server
Sunday, September 19, 2010
ZK - Open Source Ajax
http://www.zkoss.org/demo/
Apache Lucene
http://lucene.apache.org/java/docs/index.html
Apache Lucene is a high-performance, full-featured text search engine library written entirely in Java. It is a technology suitable for nearly any application that requires full-text search, especially cross-platform.
Apache Lucene
http://lucene.apache.org/java/docs/index.html
Apache Lucene is a high-performance, full-featured text search engine library written entirely in Java. It is a technology suitable for nearly any application that requires full-text search, especially cross-platform.
PCA Principal component analysis
http://ordination.okstate.edu/PCA.htm
owever, with more than three dimensions, we usually need a little help. What PCA does is that it takes your cloud of data points, and rotates it such that the maximum variability is visible. Another way of saying this is that it identifies your most important gradients.
http://en.wikipedia.org/wiki/Mahalanobis_distance
In statistics, Mahalanobis distance is a distance measure introduced by P. C. Mahalanobis in 1936.[1] It is based on correlations between variables by which different patterns can be identified and analyzed. It is a useful way of determining similarity of an unknown sample set to a known one
http://onlinelibrary.wiley.com/doi/10.1002/wics.101/abstract
Principal component analysis
Hervé Abdi1,*, Lynne J. Williams2
http://www.cs.otago.ac.nz/cosc453/student_tutorials/principal_components.pdf
http://stats.stackexchange.com/questions/2691/making-sense-of-principal-component-analysis-eigenvectors-eigenvalues
I would never try to explain this to my grandmother, but if I had to talk generally about dimension reduction techniques, I'd point to this trivial projection example (not PCA). Suppose you have a Calder mobile that is very complex. Some points in 3-d space close to each other, others aren't. If we hung this mobile from the ceiling and shined light on it from one angle, we get a projection onto a lower dimension plane (a 2-d wall). Now, if this mobile is mainly wide in one direction, but skinny in the other direction, we can rotate it to get projections that differ in usefulness. Intuitively, a skinny shape in one dimension projected on a wall is less useful - all the shadows overlap and don't give us much information. However, if we rotate it so the light shines on the wide side, we get a better picture of the reduced dimension data - points are more spread out. This is often what we want. I think my grandmother could understand that :-)
owever, with more than three dimensions, we usually need a little help. What PCA does is that it takes your cloud of data points, and rotates it such that the maximum variability is visible. Another way of saying this is that it identifies your most important gradients.
http://en.wikipedia.org/wiki/Mahalanobis_distance
In statistics, Mahalanobis distance is a distance measure introduced by P. C. Mahalanobis in 1936.[1] It is based on correlations between variables by which different patterns can be identified and analyzed. It is a useful way of determining similarity of an unknown sample set to a known one
http://onlinelibrary.wiley.com/doi/10.1002/wics.101/abstract
Principal component analysis
Hervé Abdi1,*, Lynne J. Williams2
http://www.cs.otago.ac.nz/cosc453/student_tutorials/principal_components.pdf
http://stats.stackexchange.com/questions/2691/making-sense-of-principal-component-analysis-eigenvectors-eigenvalues
I would never try to explain this to my grandmother, but if I had to talk generally about dimension reduction techniques, I'd point to this trivial projection example (not PCA). Suppose you have a Calder mobile that is very complex. Some points in 3-d space close to each other, others aren't. If we hung this mobile from the ceiling and shined light on it from one angle, we get a projection onto a lower dimension plane (a 2-d wall). Now, if this mobile is mainly wide in one direction, but skinny in the other direction, we can rotate it to get projections that differ in usefulness. Intuitively, a skinny shape in one dimension projected on a wall is less useful - all the shadows overlap and don't give us much information. However, if we rotate it so the light shines on the wide side, we get a better picture of the reduced dimension data - points are more spread out. This is often what we want. I think my grandmother could understand that :-)
GWAS database
http://www.biomedcentral.com/1471-2350/10/6
An Open Access Database of Genome-wide Association Results
Andrew D Johnson1,2 email and Christopher J O'Donnell1,2,3 email
1 National Heart, Lung, and Blood Institute's Framingham Heart Study, Framingham, MA, USA
2 Division of Intramural Research, National Heart, Lung and Blood Institute, Bethesda, MD, USA
3 Cardiology Division, Department of Medicine, Massachusetts General Hospital, Boston, MA, USA
author email corresponding author email
BMC Medical Genetics 2009, 10:6doi:10.1186/1471-2350-10-6
dbSNP
http://www.ncbi.nlm.nih.gov/projects/SNP/
http://hugenavigator.net
https://gwas.lifesciencedb.jp/cgi-bin/gwasdb/gwas_top.cgi
dbGaP - genotype and phenotype
http://www.ncbi.nlm.nih.gov/sites/entrez?Db=gap
http://www.stats.ox.ac.uk/~marchini/software/gwas/gwas.html
http://www.illumina.com/applications/gwas.ilmn
Several recent reviews highlight the need for new methods
(Thornton-Wells et al., 2004) and discuss and compare different
strategies for detecting statistical epistasis (Cordell, 2009; Motsinger
et al., 2007). The methods reviewed by Cordell (2009) include
novel approaches such as combinatorial partitioning (Culverhouse
et al., 2004; Nelson et al., 2001) and logic regression (Kooperberg
et al., 2001; Kooperberg and Ruczinski, 2005) and machine learning
approaches such as random forests (RFs). Below, we briefly
review two of these methods, RFs and multifactor dimensionality
reduction (MDR) that have been developed to address these issues.
An Open Access Database of Genome-wide Association Results
Andrew D Johnson1,2 email and Christopher J O'Donnell1,2,3 email
1 National Heart, Lung, and Blood Institute's Framingham Heart Study, Framingham, MA, USA
2 Division of Intramural Research, National Heart, Lung and Blood Institute, Bethesda, MD, USA
3 Cardiology Division, Department of Medicine, Massachusetts General Hospital, Boston, MA, USA
author email corresponding author email
BMC Medical Genetics 2009, 10:6doi:10.1186/1471-2350-10-6
dbSNP
http://www.ncbi.nlm.nih.gov/projects/SNP/
http://hugenavigator.net
https://gwas.lifesciencedb.jp/cgi-bin/gwasdb/gwas_top.cgi
dbGaP - genotype and phenotype
http://www.ncbi.nlm.nih.gov/sites/entrez?Db=gap
http://www.stats.ox.ac.uk/~marchini/software/gwas/gwas.html
http://www.illumina.com/applications/gwas.ilmn
Several recent reviews highlight the need for new methods
(Thornton-Wells et al., 2004) and discuss and compare different
strategies for detecting statistical epistasis (Cordell, 2009; Motsinger
et al., 2007). The methods reviewed by Cordell (2009) include
novel approaches such as combinatorial partitioning (Culverhouse
et al., 2004; Nelson et al., 2001) and logic regression (Kooperberg
et al., 2001; Kooperberg and Ruczinski, 2005) and machine learning
approaches such as random forests (RFs). Below, we briefly
review two of these methods, RFs and multifactor dimensionality
reduction (MDR) that have been developed to address these issues.
The Challenge of Information
http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=nap9990&part=a20003171ddd0000015
Gene ID mapping conversion - BioMart, DAVID
http://biostar.stackexchange.com/questions/22/gene-id-conversion-tool
- DAVID (http://david.abcc.ncifcrf.gov/conversion.jsp)
- BioMart http://www.biomedcentral.com/1471-2164/10/22
- DAVID (http://david.abcc.ncifcrf.gov/conversion.jsp)
- BioMart http://www.biomedcentral.com/1471-2164/10/22
BioMart enables scientists to perform advanced querying of biological data sources through a single web interface. The power of the system comes from integrated querying of data sources regardless of their geographical locations. Once these queries have been defined, they may be automated with its "scripting at the click of a button" functionality. BioMart's capabilities are extended by integration with several widely used software packages such as BioConductor, DAS, Galaxy, Cytoscape, Taverna. In this paper, we describe all aspects of BioMart from a user's perspective and demonstrate how it can be used to solve real biological use cases such as SNP selection for candidate gene screening or annotation of microarray results.
Wordle and WordCram about "word clouds"
http://www.wordle.net/
Wordle is a toy for generating “word clouds” from text that you provide. The clouds give greater prominence to words that appear more frequently in the source text.
http://wordcram.wordpress.com/
WordCram is customizable, but you have to know how it works, what each piece does, to get what you’re after.
The whole idea of a word cloud is to draw a bunch of words according to their weight. With WordCram, you hand it the weighted words and your Processing sketch, and it’ll generate the word cloud image for you.
Wordle is a toy for generating “word clouds” from text that you provide. The clouds give greater prominence to words that appear more frequently in the source text.
http://wordcram.wordpress.com/
WordCram is customizable, but you have to know how it works, what each piece does, to get what you’re after.
The whole idea of a word cloud is to draw a bunch of words according to their weight. With WordCram, you hand it the weighted words and your Processing sketch, and it’ll generate the word cloud image for you.
Google Insights for Search, Google Trends
Google Insights (friend of Google Trends) for Search
Is useful in viewing trends of search keywords, trends such as frequency, location, and time.
The horizontal axis of the main graph represents time (starting from some time in 2004), and the vertical is how often a term is searched for relative to the total number of searches, globally.
http://en.wikipedia.org/wiki/Google_Insights_for_Search
http://en.wikipedia.org/wiki/Google_Trends
http://www.google.com/support/insights//bin/answer.py?hl=en-US&answer=96693
http://www.google.com/intl/en/trends/about.html
Python Google Trends Information Retrieval Quasi-API
pyGTrends 0.8
http://pypi.python.org/pypi/pyGTrends/0.8
http://zoastertech.com/projects/googletrends/?page=Third+party+API+Tools
http://www.juiceanalytics.com/openjuice/programmatic-google-trends-api/
http://bradjasper.com/blog/use-python-to-access-google-insights-api.html
Google Search API for python
A useful Python API to parse Google search results
http://blackcodeseo.com/google-search-api-for-python/
BeautifulSoup, Mechanize
Papers
http://www.ncbi.nlm.nih.gov/pubmed/20679285
J Public Health (Oxf). 2010 Aug 5. [Epub ahead of print]
Media coverage and public reaction to a celebrity cancer diagnosis.
Metcalfe D, Price C, Powell J.
Institute of Clinical Education, Warwick Medical School, Coventry CV4 7AL, UK.
http://www.nature.com/nature/journal/v457/n7232/full/nature07634.html
Nature 457, 1012-1014 (19 February 2009) | doi:10.1038/nature07634; Received 14 August 2008; Accepted 13 November 2008; Published online 19 November 2008; Corrected 19 February 2009
Detecting influenza epidemics using search engine query data
Jeremy Ginsberg1, Matthew H. Mohebbi1, Rajan S. Patel1, Lynnette Brammer2, Mark S. Smolinski1 & Larry Brilliant1
Books
- Plant Growth and Development: A Molecular Approach.
(1994). Donald Fosket, Aademic Press.
- Plant Physiology by Taiz, L. and Zeiger, E., latest edition, Sinauer Associates Inc.
http://www.sfu.ca/biology/courses/outlines/1067/366.htm
http://www.google.ca/url?sa=t&rct=j&q=stages+of+seed+development+histodifferentiation&source=web&cd=1&ved=0CCQQFjAA&url=http%3A%2F%2Fwww.psla.umd.edu%2Ffaculty%2FColeman%2Fseed%2520structure%2520and%2520development.pdf&ei=KDEoT57cIsqoiQK6ieGQAQ&usg=AFQjCNHR6RVowlDYbqsqp5cA0TYPBjMfBw&sig2=F8-lOKj_g2sTz5QDZP0-CA
Is useful in viewing trends of search keywords, trends such as frequency, location, and time.
The horizontal axis of the main graph represents time (starting from some time in 2004), and the vertical is how often a term is searched for relative to the total number of searches, globally.
http://en.wikipedia.org/wiki/Google_Insights_for_Search
http://en.wikipedia.org/wiki/Google_Trends
http://www.google.com/support/insights//bin/answer.py?hl=en-US&answer=96693
http://www.google.com/intl/en/trends/about.html
Python Google Trends Information Retrieval Quasi-API
pyGTrends 0.8
http://pypi.python.org/pypi/pyGTrends/0.8
http://zoastertech.com/projects/googletrends/?page=Third+party+API+Tools
http://www.juiceanalytics.com/openjuice/programmatic-google-trends-api/
http://bradjasper.com/blog/use-python-to-access-google-insights-api.html
Google Search API for python
A useful Python API to parse Google search results
http://blackcodeseo.com/google-search-api-for-python/
BeautifulSoup, Mechanize
Papers
http://www.ncbi.nlm.nih.gov/pubmed/20679285
J Public Health (Oxf). 2010 Aug 5. [Epub ahead of print]
Media coverage and public reaction to a celebrity cancer diagnosis.
Metcalfe D, Price C, Powell J.
Institute of Clinical Education, Warwick Medical School, Coventry CV4 7AL, UK.
http://www.nature.com/nature/journal/v457/n7232/full/nature07634.html
Nature 457, 1012-1014 (19 February 2009) | doi:10.1038/nature07634; Received 14 August 2008; Accepted 13 November 2008; Published online 19 November 2008; Corrected 19 February 2009
Detecting influenza epidemics using search engine query data
Jeremy Ginsberg1, Matthew H. Mohebbi1, Rajan S. Patel1, Lynnette Brammer2, Mark S. Smolinski1 & Larry Brilliant1
Books
- Plant Growth and Development: A Molecular Approach.
(1994). Donald Fosket, Aademic Press.
- Plant Physiology by Taiz, L. and Zeiger, E., latest edition, Sinauer Associates Inc.
http://www.sfu.ca/biology/courses/outlines/1067/366.htm
http://www.google.ca/url?sa=t&rct=j&q=stages+of+seed+development+histodifferentiation&source=web&cd=1&ved=0CCQQFjAA&url=http%3A%2F%2Fwww.psla.umd.edu%2Ffaculty%2FColeman%2Fseed%2520structure%2520and%2520development.pdf&ei=KDEoT57cIsqoiQK6ieGQAQ&usg=AFQjCNHR6RVowlDYbqsqp5cA0TYPBjMfBw&sig2=F8-lOKj_g2sTz5QDZP0-CA
Friday, September 17, 2010
Rheumatoid arthritis (RA)
http://www.articlesbase.com/medicine-articles/is-rheumatoid-arthritis-a-genetic-disease-235438.html
Rheumatoid arthritis (RA) is the most common inflammatory type of arthritis. It affects more than 2 million Americans and is still a leading cause of both disability as well as days lost from work. RA is more common in women than men, and typical onset for the disease is between 25 and 50 years of age. Symptoms of rheumatoid arthritis include swelling, loss of movement, stiffness, and pain in joints, most commonly, the fingers and wrists.
RA is to be feared and respected because it is a systemic autoimmune chronic condition that affects internal organs as well as joints. While the cause of RA is still not completely known, a recent study published in the New England Journal of Medicine conducted by researchers in the United States and Sweden links a genetic region to rheumatoid arthritis.
Read more: http://www.articlesbase.com/medicine-articles/is-rheumatoid-arthritis-a-genetic-disease-235438.html#ixzz0zrFG5cDa
Under Creative Commons License: Attribution
Both groups of researchers used the new genome-wide association approach, which allows researchers to examine 300,000 to 500,000 small discrepancies in genetic material. Researchers examined genetic material in blood samples of all individuals that were part of the study.
The researchers found two genes in chromosome 9 responsible for the inflammation associated with RA: TRAF1 and C5. TRAF 1 codes for tumor necrosis factor, a specific target for many of the new biologic drugs used to treat RA. C5 codes for complement, a protein that also plays a big role in inflammation. Other genetic predisposing factors have been identified previously. These include HLA-DRB1 and PTPN22.
Elaine F. Remmers, Ph.D. in the Genetics and Genonics Branch of the NAMS Intramural Research Program and one of the authors of this study stated in a press release, "TRAF1-C5 showed association not only in the sample that we did with the North American Rheumatoid Arthritis Consortium but also independently in the Swedish group. By combining our information, we were able to make a much stronger case. The combined evidence was pretty impressive."
The researchers are unclear as to both how these genes are connected to RA as well as to which gene is causing the condition. Remmers added, "Actually, both genes are very interesting candidates. They both control inflammatory processes that really are relevant for the disease, so we could easily envision either of them playing a role - or both."
Read more: http://www.articlesbase.com/medicine-articles/is-rheumatoid-arthritis-a-genetic-disease-235438.html#ixzz0zrFbqI1M
Under Creative Commons License: Attribution
The authors hope that future research can reveal more about how these genes are linked to RA. They also hope that by learning more about the genes' connection to the disease, they will become closer to producing more effective treatment for the condition.
Remmers went on to say, "We are hoping that we will find variants in either of the genes that will lead us to new targets for therapy. Once we understand how the RA-associated variants work, we may be able interfere with the pathways the variants are influencing and either prevent the disease or block its progression."
Read more: http://www.articlesbase.com/medicine-articles/is-rheumatoid-arthritis-a-genetic-disease-235438.html#ixzz0zrFh2Bvx
Under Creative Commons License: Attribution
http://www.physicaltherapy.med.ubc.ca/faculty_staff/faculty_staff_directory/faculty_directory_update/Linda_Li/ANSWER_Information.htm
N Engl J Med. 2007 Sep 20;357(12):1199-209. Epub 2007 Sep 5.
TRAF1-C5 as a risk locus for rheumatoid arthritis--a genomewide study.
Plenge RM, Seielstad M, Padyukov L, Lee AT, Remmers EF, Ding B, Liew A, Khalili H, Chandrasekaran A, Davies LR, Li W, Tan AK, Bonnard C, Ong RT, Thalamuthu A, Pettersson S, Liu C, Tian C, Chen WV, Carulli JP, Beckman EM, Altshuler D, Alfredsson L, Criswell LA, Amos CI, Seldin MF, Kastner DL, Klareskog L, Gregersen PK.
Broad Institute of Harvard and the Massachusetts Institute of Technology, Cambridge, MA, USA.
Comment in:
* N Engl J Med. 2007 Sep 20;357(12):1250-1.
http://www.ncbi.nlm.nih.gov/pubmed/17804836
http://www.wrongdiagnosis.com/r/rheumatoid_arthritis/stats.htm
The following statistics relate to the prevalence of Rheumatoid arthritis:
* 2.1 million people with rheumatoid arthritis (NIAMS)
* 1,736,099 people with rheumatoid arthritis in the USA 1996 1
* 2.1 million cases of RA in the US (NIH, The National Women’s Health Centre, 2004)
* 2 to 3 times more common in women than men in the US (NIH, The National Women’s Health Centre, 2004)
* About 2.5 million people in the US (American Medical Women’s Association)
* Affects women 3 times more than men in the US (American Medical Women’s Association)
http://www.cdc.gov/arthritis/data_statistics/arthritis_related_stats.htm
http://www.creakyjoints.com/go/article0062.shtml
general response to an emphasis on genetic determinism is.
Genotyping is used to identify candidate gene regions for genetic studies via genome-wide association studies (generally 10k to 500k markers), genome-wide linkage studies (6k markers) plus fine mapping panels, and custom candidate gene approaches assaying 96 to 1536 SNPs at one time.
http://www.clip.ubc.ca/research.shtm
Rheumatoid arthritis (RA) is the most common inflammatory type of arthritis. It affects more than 2 million Americans and is still a leading cause of both disability as well as days lost from work. RA is more common in women than men, and typical onset for the disease is between 25 and 50 years of age. Symptoms of rheumatoid arthritis include swelling, loss of movement, stiffness, and pain in joints, most commonly, the fingers and wrists.
RA is to be feared and respected because it is a systemic autoimmune chronic condition that affects internal organs as well as joints. While the cause of RA is still not completely known, a recent study published in the New England Journal of Medicine conducted by researchers in the United States and Sweden links a genetic region to rheumatoid arthritis.
Read more: http://www.articlesbase.com/medicine-articles/is-rheumatoid-arthritis-a-genetic-disease-235438.html#ixzz0zrFG5cDa
Under Creative Commons License: Attribution
Both groups of researchers used the new genome-wide association approach, which allows researchers to examine 300,000 to 500,000 small discrepancies in genetic material. Researchers examined genetic material in blood samples of all individuals that were part of the study.
The researchers found two genes in chromosome 9 responsible for the inflammation associated with RA: TRAF1 and C5. TRAF 1 codes for tumor necrosis factor, a specific target for many of the new biologic drugs used to treat RA. C5 codes for complement, a protein that also plays a big role in inflammation. Other genetic predisposing factors have been identified previously. These include HLA-DRB1 and PTPN22.
Elaine F. Remmers, Ph.D. in the Genetics and Genonics Branch of the NAMS Intramural Research Program and one of the authors of this study stated in a press release, "TRAF1-C5 showed association not only in the sample that we did with the North American Rheumatoid Arthritis Consortium but also independently in the Swedish group. By combining our information, we were able to make a much stronger case. The combined evidence was pretty impressive."
The researchers are unclear as to both how these genes are connected to RA as well as to which gene is causing the condition. Remmers added, "Actually, both genes are very interesting candidates. They both control inflammatory processes that really are relevant for the disease, so we could easily envision either of them playing a role - or both."
Read more: http://www.articlesbase.com/medicine-articles/is-rheumatoid-arthritis-a-genetic-disease-235438.html#ixzz0zrFbqI1M
Under Creative Commons License: Attribution
The authors hope that future research can reveal more about how these genes are linked to RA. They also hope that by learning more about the genes' connection to the disease, they will become closer to producing more effective treatment for the condition.
Remmers went on to say, "We are hoping that we will find variants in either of the genes that will lead us to new targets for therapy. Once we understand how the RA-associated variants work, we may be able interfere with the pathways the variants are influencing and either prevent the disease or block its progression."
Read more: http://www.articlesbase.com/medicine-articles/is-rheumatoid-arthritis-a-genetic-disease-235438.html#ixzz0zrFh2Bvx
Under Creative Commons License: Attribution
http://www.physicaltherapy.med.ubc.ca/faculty_staff/faculty_staff_directory/faculty_directory_update/Linda_Li/ANSWER_Information.htm
N Engl J Med. 2007 Sep 20;357(12):1199-209. Epub 2007 Sep 5.
TRAF1-C5 as a risk locus for rheumatoid arthritis--a genomewide study.
Plenge RM, Seielstad M, Padyukov L, Lee AT, Remmers EF, Ding B, Liew A, Khalili H, Chandrasekaran A, Davies LR, Li W, Tan AK, Bonnard C, Ong RT, Thalamuthu A, Pettersson S, Liu C, Tian C, Chen WV, Carulli JP, Beckman EM, Altshuler D, Alfredsson L, Criswell LA, Amos CI, Seldin MF, Kastner DL, Klareskog L, Gregersen PK.
Broad Institute of Harvard and the Massachusetts Institute of Technology, Cambridge, MA, USA.
Comment in:
* N Engl J Med. 2007 Sep 20;357(12):1250-1.
http://www.ncbi.nlm.nih.gov/pubmed/17804836
http://www.wrongdiagnosis.com/r/rheumatoid_arthritis/stats.htm
The following statistics relate to the prevalence of Rheumatoid arthritis:
* 2.1 million people with rheumatoid arthritis (NIAMS)
* 1,736,099 people with rheumatoid arthritis in the USA 1996 1
* 2.1 million cases of RA in the US (NIH, The National Women’s Health Centre, 2004)
* 2 to 3 times more common in women than men in the US (NIH, The National Women’s Health Centre, 2004)
* About 2.5 million people in the US (American Medical Women’s Association)
* Affects women 3 times more than men in the US (American Medical Women’s Association)
http://www.cdc.gov/arthritis/data_statistics/arthritis_related_stats.htm
http://www.creakyjoints.com/go/article0062.shtml
general response to an emphasis on genetic determinism is.
Genotyping is used to identify candidate gene regions for genetic studies via genome-wide association studies (generally 10k to 500k markers), genome-wide linkage studies (6k markers) plus fine mapping panels, and custom candidate gene approaches assaying 96 to 1536 SNPs at one time.
http://www.clip.ubc.ca/research.shtm
autoimmunity
Autoimmunity is the failure of an organism to recognize its own constituent parts as self, which allows an immune response against its own cells and tissues. Any disease that results from such an aberrant immune response is termed an autoimmune disease. Autoimmunity is often caused by a lack of germ development of a target body and as such the immune response acts against its own cells and tissues (Flowers 2009). Prominent examples include Coeliac disease, diabetes mellitus type 1 (IDDM), systemic lupus erythematosus (SLE), Sjögren's syndrome, Churg-Strauss Syndrome, Hashimoto's thyroiditis, Graves' disease, idiopathic thrombocytopenic purpura, and rheumatoid arthritis (RA). See List of autoimmune diseases.
http://en.wikipedia.org/wiki/Autoimmunity
http://en.wikipedia.org/wiki/Autoimmunity
Protein–protein interaction and pathway databases, a graphical review
1. Tomas Klingström
Tomas S. Klingström is studying at the Master of Science programme in Molecular Biotechnology Engineering at Uppsala University (estimated to finish in January 2011) and recipient of the Anders Wall scholarship to young researchers 2010. His main area of interest is large-scale studies of protein–protein interactions on a proteomic level for usage in systems biology and the pharmaceutical industry.
and
2. Dariusz Plewczynski
Dariusz Plewczynski is a Research Professor at University of Warsaw in Interdyscyplinary Centre for Mathematical and Computational Modelling, Poland. His main expertise covers protein interactions (including both protein–ligand and protein–protein) especially in the context of docking and high-throughput studies. He applies machine learning algorithms and clustering techniques for functional annotation of protein sequences and structures.
1. Corresponding author. Dariusz Plewczynski, Interdisciplinary Centre for Mathematical and Computational Modelling, University of Warsaw, Pawinskiego 5a Street, 02-106 Warsaw, Poland. Tel: +4 822 554 0838 and +4 850 472 6203; Fax: +4 822 554 0801; E-mail: darman@icm.edu.pl
* Received July 13, 2010.
* Accepted August 14, 2010.
http://bib.oxfordjournals.org/content/early/2010/09/16/bib.bbq064
Tomas S. Klingström is studying at the Master of Science programme in Molecular Biotechnology Engineering at Uppsala University (estimated to finish in January 2011) and recipient of the Anders Wall scholarship to young researchers 2010. His main area of interest is large-scale studies of protein–protein interactions on a proteomic level for usage in systems biology and the pharmaceutical industry.
and
2. Dariusz Plewczynski
Dariusz Plewczynski is a Research Professor at University of Warsaw in Interdyscyplinary Centre for Mathematical and Computational Modelling, Poland. His main expertise covers protein interactions (including both protein–ligand and protein–protein) especially in the context of docking and high-throughput studies. He applies machine learning algorithms and clustering techniques for functional annotation of protein sequences and structures.
1. Corresponding author. Dariusz Plewczynski, Interdisciplinary Centre for Mathematical and Computational Modelling, University of Warsaw, Pawinskiego 5a Street, 02-106 Warsaw, Poland. Tel: +4 822 554 0838 and +4 850 472 6203; Fax: +4 822 554 0801; E-mail: darman@icm.edu.pl
* Received July 13, 2010.
* Accepted August 14, 2010.
http://bib.oxfordjournals.org/content/early/2010/09/16/bib.bbq064
Research About - Genetics
http://www.cihr-irsc.gc.ca/e/39999.html
http://www.cihr-irsc.gc.ca/e/40613.html
The Canadian Institutes of Health Research (CIHR) is the Government of Canada's agency for health research. CIHR's mission is to create new scientific knowledge and to catalyze its translation into improved health, more effective health services and products, and a strengthened Canadian health-care system. Composed of 13 Institutes, CIHR provides leadership and support to more than 13,000 health researchers and trainees across Canada. Through CIHR, the Government of Canada invested approximately $244.1 million in 2008-09 in genetics-related research.
CIHR's Institute of Genetics (CIHR-IG) has identified six themes as research priorities for Canada: integrating the physical and applied sciences into health research; proteomics and bioinformatics; genomic medicine; population genetics, genetic epidemiology and complex diseases; health services for genetic diseases; and genetics and ethical, legal and social issues. To learn more about these priorities and other CIHR-IG activities, please visit the Institute's website.
"Some people age in a healthy fashion despite many physical health challenges, while others who are in good physical health age less optimally. What explains this phenomenon? The study will answer questions that are relevant to decision-makers to improve the health of Canadians," said Dr. Parminder Raina, McMaster University, who is leading the study along with Dr. Christina Wolfson, McGill University, and Dr. Susan Kirkland, Dalhousie University.
New Online Resource Available for People with Rheumatoid Arthritis
Vancouver: With support from CIHR, researchers at the University of British Columbia and the Arthritis Research Centre of Canada are creating an interactive tool that will help patients with rheumatoid arthritis find the best treatment option for their condition. Led by clinical epidemiologist Dr. Linda Li and social worker Paul Adam, the team is working with the Centre for Digital Media in Vancouver to create the Animated, Self-serve, Web-based Research (ANSWER) tool. "The patients can use the program to find information tailored to their condition and print out a one-page report with their concerns, questions and initial decisions about treatment. They can then take this report to discuss with their doctor," says Dr. Li.
Universal Flu Shots Reduce Antibiotic Use
Vancouver: A program of universal vaccination for seasonal flu sharply reduced inappropriate antibiotic use, a CIHR-supported study has found. Dr. Fawziah Marra at the University of British Columbia and BC Centre for Disease Control discovered that the universal influenza program, which began in Ontario in 2000, reduced the use of emergency services as well as the influenza-associated mortality rate. In addition, the universal program also resulted in 64% fewer flu-associated antibiotic prescriptions. In contrast, the rate of such prescriptions did not change significantly in other Canadian provinces, where flu vaccination was targeted to specific groups, Dr. Marra and her team said. "Jurisdictions wishing to decrease antibiotic use might consider programs to increase influenza vaccination," the researchers concluded.
http://www.cihr-irsc.gc.ca/e/40613.html
The Canadian Institutes of Health Research (CIHR) is the Government of Canada's agency for health research. CIHR's mission is to create new scientific knowledge and to catalyze its translation into improved health, more effective health services and products, and a strengthened Canadian health-care system. Composed of 13 Institutes, CIHR provides leadership and support to more than 13,000 health researchers and trainees across Canada. Through CIHR, the Government of Canada invested approximately $244.1 million in 2008-09 in genetics-related research.
CIHR's Institute of Genetics (CIHR-IG) has identified six themes as research priorities for Canada: integrating the physical and applied sciences into health research; proteomics and bioinformatics; genomic medicine; population genetics, genetic epidemiology and complex diseases; health services for genetic diseases; and genetics and ethical, legal and social issues. To learn more about these priorities and other CIHR-IG activities, please visit the Institute's website.
"Some people age in a healthy fashion despite many physical health challenges, while others who are in good physical health age less optimally. What explains this phenomenon? The study will answer questions that are relevant to decision-makers to improve the health of Canadians," said Dr. Parminder Raina, McMaster University, who is leading the study along with Dr. Christina Wolfson, McGill University, and Dr. Susan Kirkland, Dalhousie University.
New Online Resource Available for People with Rheumatoid Arthritis
Vancouver: With support from CIHR, researchers at the University of British Columbia and the Arthritis Research Centre of Canada are creating an interactive tool that will help patients with rheumatoid arthritis find the best treatment option for their condition. Led by clinical epidemiologist Dr. Linda Li and social worker Paul Adam, the team is working with the Centre for Digital Media in Vancouver to create the Animated, Self-serve, Web-based Research (ANSWER) tool. "The patients can use the program to find information tailored to their condition and print out a one-page report with their concerns, questions and initial decisions about treatment. They can then take this report to discuss with their doctor," says Dr. Li.
Universal Flu Shots Reduce Antibiotic Use
Vancouver: A program of universal vaccination for seasonal flu sharply reduced inappropriate antibiotic use, a CIHR-supported study has found. Dr. Fawziah Marra at the University of British Columbia and BC Centre for Disease Control discovered that the universal influenza program, which began in Ontario in 2000, reduced the use of emergency services as well as the influenza-associated mortality rate. In addition, the universal program also resulted in 64% fewer flu-associated antibiotic prescriptions. In contrast, the rate of such prescriptions did not change significantly in other Canadian provinces, where flu vaccination was targeted to specific groups, Dr. Marra and her team said. "Jurisdictions wishing to decrease antibiotic use might consider programs to increase influenza vaccination," the researchers concluded.
Bioinformatics challenges for genome-wide association studies
http://bioinformatics.oxfordjournals.org/content/26/4/445.abstract
Abstract
Motivation: The sequencing of the human genome has made it possible to identify an informative set of >1 million single nucleotide polymorphisms (SNPs) across the genome that can be used to carry out genome-wide association studies (GWASs). The availability of massive amounts of GWAS data has necessitated the development of new biostatistical methods for quality control, imputation and analysis issues including multiple testing. This work has been successful and has enabled the discovery of new associations that have been replicated in multiple studies. However, it is now recognized that most SNPs discovered via GWAS have small effects on disease susceptibility and thus may not be suitable for improving health care through genetic testing. One likely explanation for the mixed results of GWAS is that the current biostatistical analysis paradigm is by design agnostic or unbiased in that it ignores all prior knowledge about disease pathobiology. Further, the linear modeling framework that is employed in GWAS often considers only one SNP at a time thus ignoring their genomic and environmental context. There is now a shift away from the biostatistical approach toward a more holistic approach that recognizes the complexity of the genotype–phenotype relationship that is characterized by significant heterogeneity and gene–gene and gene–environment interaction. We argue here that bioinformatics has an important role to play in addressing the complexity of the underlying genetic basis of common human diseases. The goal of this review is to identify and discuss those GWAS challenges that will require computational methods.
Abstract
Motivation: The sequencing of the human genome has made it possible to identify an informative set of >1 million single nucleotide polymorphisms (SNPs) across the genome that can be used to carry out genome-wide association studies (GWASs). The availability of massive amounts of GWAS data has necessitated the development of new biostatistical methods for quality control, imputation and analysis issues including multiple testing. This work has been successful and has enabled the discovery of new associations that have been replicated in multiple studies. However, it is now recognized that most SNPs discovered via GWAS have small effects on disease susceptibility and thus may not be suitable for improving health care through genetic testing. One likely explanation for the mixed results of GWAS is that the current biostatistical analysis paradigm is by design agnostic or unbiased in that it ignores all prior knowledge about disease pathobiology. Further, the linear modeling framework that is employed in GWAS often considers only one SNP at a time thus ignoring their genomic and environmental context. There is now a shift away from the biostatistical approach toward a more holistic approach that recognizes the complexity of the genotype–phenotype relationship that is characterized by significant heterogeneity and gene–gene and gene–environment interaction. We argue here that bioinformatics has an important role to play in addressing the complexity of the underlying genetic basis of common human diseases. The goal of this review is to identify and discuss those GWAS challenges that will require computational methods.
Ten years of genetics and genomics: what have we achieved and where are we heading
http://www.nature.com/nrg/journal/v11/n10/full/nrg2878.html
Nature Reviews Genetics 11, 723-733 (October 2010) | doi:10.1038/nrg2878
Viewpoint: Ten years of genetics and genomics: what have we achieved and where are we heading?
genome wide association, epigenetics, miRNA, ncRNA (non-coding RNA)
http://www.cihr-irsc.gc.ca/e/39999.html
Nature Reviews Genetics 11, 723-733 (October 2010) | doi:10.1038/nrg2878
Viewpoint: Ten years of genetics and genomics: what have we achieved and where are we heading?
genome wide association, epigenetics, miRNA, ncRNA (non-coding RNA)
http://www.cihr-irsc.gc.ca/e/39999.html
Epigenomics
http://nihroadmap.nih.gov/epigenomics/
Epigenetics is an emerging frontier of science that involves the study of changes in the regulation of gene activity and expression that are not dependent on gene sequence. For purposes of this program, epigenetics refers to both heritable changes in gene activity and expression (in the progeny of cells or of individuals) and also stable, long-term alterations in the transcriptional potential of a cell that are not necessarily heritable. While epigenetics refers to the study of single genes or sets of genes, epigenomics refers to more global analyses of epigenetic changes across the entire genome.
Epigenetics is an emerging frontier of science that involves the study of changes in the regulation of gene activity and expression that are not dependent on gene sequence. For purposes of this program, epigenetics refers to both heritable changes in gene activity and expression (in the progeny of cells or of individuals) and also stable, long-term alterations in the transcriptional potential of a cell that are not necessarily heritable. While epigenetics refers to the study of single genes or sets of genes, epigenomics refers to more global analyses of epigenetic changes across the entire genome.
Metabolome
Metabolome[1] refers to the complete set of small-molecule metabolites (such as metabolic intermediates, hormones and other signalling molecules, and secondary metabolites) to be found within a biological sample, such as a single organism. The word was coined in analogy with transcriptomics and proteomics; like the transcriptome and the proteome, the metabolome is dynamic, changing from second to second.
genome-wide association study (GWAS)
http://www.genome.gov/20019523
What is a genome-wide association study (GWAS)?
A genome-wide association study is an approach that involves rapidly scanning markers across the complete sets of DNA, or genomes, of many people to find genetic variations associated with a particular disease. Once new genetic associations are identified, researchers can use the information to develop better strategies to detect, treat and prevent the disease. Such studies are particularly useful in finding genetic variations that contribute to common, complex diseases, such as asthma, cancer, diabetes, heart disease and mental illnesses.
The International HapMap Project is an organization whose goal is to develop a haplotype map (HapMap) of the human genome, which will describe the common patterns of human genetic variation. The HapMap is expected to be a key resource for researchers to find genetic variants affecting health, disease and responses to drugs and environmental factors. The information produced by the project is made freely available to researchers around the world.
http://www.genesandenvironment.nih.gov/genetics/
What is a genome-wide association study (GWAS)?
A genome-wide association study is an approach that involves rapidly scanning markers across the complete sets of DNA, or genomes, of many people to find genetic variations associated with a particular disease. Once new genetic associations are identified, researchers can use the information to develop better strategies to detect, treat and prevent the disease. Such studies are particularly useful in finding genetic variations that contribute to common, complex diseases, such as asthma, cancer, diabetes, heart disease and mental illnesses.
The International HapMap Project is an organization whose goal is to develop a haplotype map (HapMap) of the human genome, which will describe the common patterns of human genetic variation. The HapMap is expected to be a key resource for researchers to find genetic variants affecting health, disease and responses to drugs and environmental factors. The information produced by the project is made freely available to researchers around the world.
http://www.genesandenvironment.nih.gov/genetics/
Gene knockdown
Gene knockdown refers to techniques by which the expression of one or more of an organism's genes is reduced, either through genetic modification (a change in the DNA of one of the organism's chromosomes) or by treatment with a reagent such as a short DNA or RNA oligonucleotide with a sequence complementary to either an mRNA transcript or a gene. If genetic modification of DNA is done, the result is a "knockdown organism". If the change in gene expression is caused by an oligonucleotide binding to an mRNA or temporarily binding to a gene, this results in a temporary change in gene expression without modification of the chromosomal DNA and is referred to as a "transient knockdown".
problem with transient knockdown is that there might be functional redundancy in the pathways so even if one gene was knocked down, other genes in the pathway might compensate for it ...
problem with transient knockdown is that there might be functional redundancy in the pathways so even if one gene was knocked down, other genes in the pathway might compensate for it ...
Google Org and Philanthropy
http://www.google.com/corporate/diversity/rise/index.html
http://www.google.org/googlers.html
http://code.google.com/edu/tools101/linux/grep.html
http://www.google.org/flutrends/
http://www.google.org/googlers.html
http://code.google.com/edu/tools101/linux/grep.html
http://www.google.org/flutrends/
Thursday, September 16, 2010
BLAT vs BLAST
http://genome.ucsc.edu/FAQ/FAQblat.html
From a practical standpoint, Blat has several advantages over BLAST:
* speed (no queues, response in seconds) at the price of lesser homology depth
* the ability to submit a long list of simultaneous queries in fasta format
* five convenient output sort options
* a direct link into the UCSC browser
* alignment block details in natural genomic order
* an option to launch the alignment later as part of a custom track
Blat is commonly used to look up the location of a sequence in the genome or determine the exon structure of an mRNA,
BLAT is not BLAST. DNA BLAT works by keeping an index of the entire genome in memory. The index consists of all non-overlapping 11-mers except for those heavily involved in repeats. The index takes up a bit less than a gigabyte of RAM. The genome itself is not kept in memory, allowing BLAT to deliver high performance on a reasonably priced Linux box. The index is used to find areas of probable homology, which are then loaded into memory for a detailed alignment. Protein BLAT works in a similar manner, except with 4-mers rather than 11-mers. The protein index takes a little more than 2 gigabytes.
BLAT was written by Jim Kent. Like most of Jim's software, interactive use on this web server is free to all. Sources and executables to run batch jobs on your own server are available free for academic, personal, and non-profit purposes. Non-exclusive commercial licenses are also available. See the Kent Informatics website for details.
From a practical standpoint, Blat has several advantages over BLAST:
* speed (no queues, response in seconds) at the price of lesser homology depth
* the ability to submit a long list of simultaneous queries in fasta format
* five convenient output sort options
* a direct link into the UCSC browser
* alignment block details in natural genomic order
* an option to launch the alignment later as part of a custom track
Blat is commonly used to look up the location of a sequence in the genome or determine the exon structure of an mRNA,
BLAT is not BLAST. DNA BLAT works by keeping an index of the entire genome in memory. The index consists of all non-overlapping 11-mers except for those heavily involved in repeats. The index takes up a bit less than a gigabyte of RAM. The genome itself is not kept in memory, allowing BLAT to deliver high performance on a reasonably priced Linux box. The index is used to find areas of probable homology, which are then loaded into memory for a detailed alignment. Protein BLAT works in a similar manner, except with 4-mers rather than 11-mers. The protein index takes a little more than 2 gigabytes.
BLAT was written by Jim Kent. Like most of Jim's software, interactive use on this web server is free to all. Sources and executables to run batch jobs on your own server are available free for academic, personal, and non-profit purposes. Non-exclusive commercial licenses are also available. See the Kent Informatics website for details.
The ENCODE Project: ENCyclopedia Of DNA Elements
http://www.genome.gov/10005107
The National Human Genome Research Institute (NHGRI) launched a public research consortium named ENCODE, the Encyclopedia Of DNA Elements, in September 2003, to carry out a project to identify all functional elements in the human genome sequence.
The second RFA, RFA HG-03-004, entitled Technologies to find functional elements in DNA, solicited applications to develop new and improved technologies for the efficient, comprehensive, high-throughput identification and verification of all types of sequence-based functional elements, particularly those other than coding sequences, for which adequate methods do not currently exist.
http://www.nature.com/nature/journal/v447/n7146/full/nature05874.html#abs
http://nar.oxfordjournals.org/content/38/suppl_1/D620.full
http://genome.ucsc.edu/ENCODE/
The National Human Genome Research Institute (NHGRI) launched a public research consortium named ENCODE, the Encyclopedia Of DNA Elements, in September 2003, to carry out a project to identify all functional elements in the human genome sequence.
The second RFA, RFA HG-03-004, entitled Technologies to find functional elements in DNA, solicited applications to develop new and improved technologies for the efficient, comprehensive, high-throughput identification and verification of all types of sequence-based functional elements, particularly those other than coding sequences, for which adequate methods do not currently exist.
http://www.nature.com/nature/journal/v447/n7146/full/nature05874.html#abs
http://nar.oxfordjournals.org/content/38/suppl_1/D620.full
http://genome.ucsc.edu/ENCODE/
Presentation
- Describe brief introduction / background info on any methods used, bayesian, methylation, machine learning, sequencing etc so audience can follow the presentation
- don't put too much words on a slide
- have a slide for major points covered and number the slides so people can refer to them after the presentation
- don't be apologetic, put a positive spin to it, you're the greatest!
- don't put too much words on a slide
- have a slide for major points covered and number the slides so people can refer to them after the presentation
- don't be apologetic, put a positive spin to it, you're the greatest!
Graduate School:
from the UBC Faculty of Graduate Studies
The Graduate
- From apprentice to expert
- Depth over breadth
- Analyzer and creator of knowledge
- Self-directed
Purpose of Masters
To understand thoroughly and think critically about what is known in a particular academic field
To learn how to conduct research in that field (and perhaps prepare for PhD study)
To begin affiliating with the academic community of the field
Purpose of Doctoral
To master the knowledge of a specific academic field – and become prepared to teach that knowledge at the university level
To make an original contribution, through research, to the knowledge within a specific field (i.e. “create new knowledge”)
To establish oneself as an expert and leader within the academic community of the field
7 Keys to Success in Graduate School
1. Be proactive – take responsibility for your own grad school experience
Think about what you really want from graduate school, and identify opportunities to attain those goals
Continue the mental transition from being told what to do, to deciding what to do
Don’t wait for faculty members to come to find you – take the initiative to build relationships
2. Establish positive relationships with your
supervisor and members of your committee
Schedule regular meetings with your entire supervisory committee – at least once a year
Have a clear purpose for each meeting, and communicate the agenda in advance to your supervisor / committee
Follow up on items discussed in meetings – keep your supervisor informed of your progress and challenges
Act as a “junior colleague” – ask questions, advance ideas, show interest and support for shared goals
3. Embrace your academic community
Seek input and collaboration from faculty members and your peers – don’t isolate yourself
Attend optional seminars and lectures within and beyond your program or department
Attend and present at conferences
Begin thinking of yourself as a member of your profession and academic field
4. Know your program requirements and timelines:
Masters students
Your program may require any or all of the following:
Coursework
Comprehensive or qualifying exams
A research thesis or major project
Public presentation and/or defense of thesis or project
UBC Graduate Policy states that:
Masters students must complete all degree requirements within 5 years of enrolment
4. Know your program requirements and timelines:
Doctoral students
Your program may require that you do coursework
Doctoral programs will require that you:
Develop and gain approval for a research proposal
Pass a comprehensive exam
Complete a research thesis (dissertation)
Defend the thesis at a Doctoral Oral Examination
UBC Policy states that Doctoral Students:
Should advance to candidacy within 2 years, and must within 3 years
Should complete all degree requirements within 5 years of
enrolment and must within 6 years
5. Create and follow an annual plan
Use UBC’s upcoming CV template as an annual guide to:
Track your specific program requirements (e.g., courses taken, comprehensives, research, thesis, etc.)
Schedule meetings with your supervisor and committee
Publish articles and produce patents, copyrights, artistic works, performances, designs, etc.
Attend conferences and make presentations
Apply for fellowships, scholarships, and research grants
Take professional development courses
6. Bring a professional approach to your studies and interactions
Key skills: organization, preparedness, collegiality, budgeting
Take workshops through the Faculty of Graduate Studies’ Professional Development Initiative (PDI) or UBC’s Teaching and Academic Growth (TAG)
Learn about research ethics and scholarly integrity
7. Seek balance and support in your life
Remember that you have friends and family outside grad school
Seek out the many resources at UBC that can help you through the tough times
Remember that this will be among the most inspiring and satisfying times in your life
And, because your mother isn’t here: “Get enough sleep, make time for physical exercise, and eat your veggies!”
Recognize that researchers before you have correlated adequate sleep, exercise, and healthy diets with critical analysis, innovation, and economy of effort
The Graduate
- From apprentice to expert
- Depth over breadth
- Analyzer and creator of knowledge
- Self-directed
Purpose of Masters
To understand thoroughly and think critically about what is known in a particular academic field
To learn how to conduct research in that field (and perhaps prepare for PhD study)
To begin affiliating with the academic community of the field
Purpose of Doctoral
To master the knowledge of a specific academic field – and become prepared to teach that knowledge at the university level
To make an original contribution, through research, to the knowledge within a specific field (i.e. “create new knowledge”)
To establish oneself as an expert and leader within the academic community of the field
7 Keys to Success in Graduate School
1. Be proactive – take responsibility for your own grad school experience
Think about what you really want from graduate school, and identify opportunities to attain those goals
Continue the mental transition from being told what to do, to deciding what to do
Don’t wait for faculty members to come to find you – take the initiative to build relationships
2. Establish positive relationships with your
supervisor and members of your committee
Schedule regular meetings with your entire supervisory committee – at least once a year
Have a clear purpose for each meeting, and communicate the agenda in advance to your supervisor / committee
Follow up on items discussed in meetings – keep your supervisor informed of your progress and challenges
Act as a “junior colleague” – ask questions, advance ideas, show interest and support for shared goals
3. Embrace your academic community
Seek input and collaboration from faculty members and your peers – don’t isolate yourself
Attend optional seminars and lectures within and beyond your program or department
Attend and present at conferences
Begin thinking of yourself as a member of your profession and academic field
4. Know your program requirements and timelines:
Masters students
Your program may require any or all of the following:
Coursework
Comprehensive or qualifying exams
A research thesis or major project
Public presentation and/or defense of thesis or project
UBC Graduate Policy states that:
Masters students must complete all degree requirements within 5 years of enrolment
4. Know your program requirements and timelines:
Doctoral students
Your program may require that you do coursework
Doctoral programs will require that you:
Develop and gain approval for a research proposal
Pass a comprehensive exam
Complete a research thesis (dissertation)
Defend the thesis at a Doctoral Oral Examination
UBC Policy states that Doctoral Students:
Should advance to candidacy within 2 years, and must within 3 years
Should complete all degree requirements within 5 years of
enrolment and must within 6 years
5. Create and follow an annual plan
Use UBC’s upcoming CV template as an annual guide to:
Track your specific program requirements (e.g., courses taken, comprehensives, research, thesis, etc.)
Schedule meetings with your supervisor and committee
Publish articles and produce patents, copyrights, artistic works, performances, designs, etc.
Attend conferences and make presentations
Apply for fellowships, scholarships, and research grants
Take professional development courses
6. Bring a professional approach to your studies and interactions
Key skills: organization, preparedness, collegiality, budgeting
Take workshops through the Faculty of Graduate Studies’ Professional Development Initiative (PDI) or UBC’s Teaching and Academic Growth (TAG)
Learn about research ethics and scholarly integrity
7. Seek balance and support in your life
Remember that you have friends and family outside grad school
Seek out the many resources at UBC that can help you through the tough times
Remember that this will be among the most inspiring and satisfying times in your life
And, because your mother isn’t here: “Get enough sleep, make time for physical exercise, and eat your veggies!”
Recognize that researchers before you have correlated adequate sleep, exercise, and healthy diets with critical analysis, innovation, and economy of effort
Graduate Student Development
http://www.grad.ubc.ca/current-students/gps-graduate-pathways-success/research-lived-experience-graduate-students
About the ALEXA-Seq data viewer
About the ALEXA-Seq data viewer
The following is a list of utilities and resources used to create the ALEXA-Seq visualization tool:
Figures and statistics: The R Project and Bioconductor
Scalable Vector Graphics was chosen as the image format: XML Graphics for the Web
General web programming: Perl
Database management systems: MySQL and Berkeley DB
Web animation java-script used to give the viewer a 'Web 2.0' style: Google AJAX APIs and Dynamic Drive
Indexing and search functionality: The Xapian Project and Omega
Web traffic analysis: Google Analytics
The following is a list of utilities and resources used to create the ALEXA-Seq visualization tool:
Figures and statistics: The R Project and Bioconductor
Scalable Vector Graphics was chosen as the image format: XML Graphics for the Web
General web programming: Perl
Database management systems: MySQL and Berkeley DB
Web animation java-script used to give the viewer a 'Web 2.0' style: Google AJAX APIs and Dynamic Drive
Indexing and search functionality: The Xapian Project and Omega
Web traffic analysis: Google Analytics
Paired-end reads in next-gen sequencing
http://seqanswers.com/forums/showthread.php?t=503
http://www.illumina.com/technology/paired_end_sequencing_assay.ilmn
ECO says ...
For example, you shear up some genomic DNA, and cut a region out at ~500bp. Then you prepare your library, and sequence 35bp from each end of each molecule. Now you have three pieces of information:
--the tag 1 sequence
--the tag 2 sequence
--that they were 500bp ± (some) apart in your genome
This gives you the ability to map to a reference (or denovo for that matter) using that distance information. It helps dramatically to resolve larger structural rearrangements (insertions, deletions, inversions), as well as helping to assemble across repetitive regions.
Structural rearrangements can be deduced when your read pairs map to a reference at a distance that is substantially different from how that library was constructed (~500bp in the above example). Let's say you had two reads that mapped to your reference 1000bp apart...this suggests there has been a deletion between those two sequence reads within your genome. Same thing with an insertion, if your reads mapped 100bp apart on the reference, this suggests that your genome has an insertion.
http://www.illumina.com/technology/paired_end_sequencing_assay.ilmn
ECO says ...
For example, you shear up some genomic DNA, and cut a region out at ~500bp. Then you prepare your library, and sequence 35bp from each end of each molecule. Now you have three pieces of information:
--the tag 1 sequence
--the tag 2 sequence
--that they were 500bp ± (some) apart in your genome
This gives you the ability to map to a reference (or denovo for that matter) using that distance information. It helps dramatically to resolve larger structural rearrangements (insertions, deletions, inversions), as well as helping to assemble across repetitive regions.
Structural rearrangements can be deduced when your read pairs map to a reference at a distance that is substantially different from how that library was constructed (~500bp in the above example). Let's say you had two reads that mapped to your reference 1000bp apart...this suggests there has been a deletion between those two sequence reads within your genome. Same thing with an insertion, if your reads mapped 100bp apart on the reference, this suggests that your genome has an insertion.
AB Solid Color Space Next-Gen sequencing
http://seqanswers.com/forums/showthread.php?t=10
Detection of a true SNP is reflected by changes in two adjacent colorspace calls, which must follow the rules above. Figure 7 below gives some examples of this principle in examining alignments.
If you’re doing the math you’ve realized there are 16 possible dinucleotides (4^2) and only 4 dyes. So data from a single color call does not tell you what base is at a given position. This is where the brilliance (and potential confusion) comes about with regard to SOLiD. There are 4 oligos for every dye, meaning there are four dinucleotides that are encoded by each dye.
Detection of a true SNP is reflected by changes in two adjacent colorspace calls, which must follow the rules above. Figure 7 below gives some examples of this principle in examining alignments.
If you’re doing the math you’ve realized there are 16 possible dinucleotides (4^2) and only 4 dyes. So data from a single color call does not tell you what base is at a given position. This is where the brilliance (and potential confusion) comes about with regard to SOLiD. There are 4 oligos for every dye, meaning there are four dinucleotides that are encoded by each dye.
RT-PCR
http://www.bio.davidson.edu/courses/immunology/Flash/RT_PCR.html
1. uses reverse transcriptase to reverse transcribe the mrna exons to cdna
2. use taq polymerase and primers to transcribe
http://pathmicro.med.sc.edu/pcr/realtime-home.htm
1. mRNA is copied to cDNA by reverse transcriptase using an oligo dT primer (random oligomers may also be used). In real-time PCR, we usually use a reverse transcriptase that has an endo H activity. This removes the mRNA allowing the second strand of DNA to be formed. A PCR mix is then set up which includes a heat-stable polymerase (such as Taq polymerase), specific primers for the gene of interest, deoxynucleotides and a suitable buffer.
pp-pcr2.gif (9025 bytes) pp-pcr3.gif (7124 bytes) 2. cDNA is denatured at more than 90 degrees (~94 degrees) so that the two strands separate. The sample is cooled to 50 to 60 degrees and specific primers are annealed that are complementary to a site on each strand. The primers sites may be up to 600 bases apart but are often about 100 bases apart, especially when real-time PCR is used.
3. The temperature is raised to 72 degrees and the heat-stable Taq DNA polymerase extends the DNA from the primers. Now we have four cDNA strands (from the original two). These are denatured again at approximately 94 degrees.
1. uses reverse transcriptase to reverse transcribe the mrna exons to cdna
2. use taq polymerase and primers to transcribe
http://pathmicro.med.sc.edu/pcr/realtime-home.htm
1. mRNA is copied to cDNA by reverse transcriptase using an oligo dT primer (random oligomers may also be used). In real-time PCR, we usually use a reverse transcriptase that has an endo H activity. This removes the mRNA allowing the second strand of DNA to be formed. A PCR mix is then set up which includes a heat-stable polymerase (such as Taq polymerase), specific primers for the gene of interest, deoxynucleotides and a suitable buffer.
pp-pcr2.gif (9025 bytes) pp-pcr3.gif (7124 bytes) 2. cDNA is denatured at more than 90 degrees (~94 degrees) so that the two strands separate. The sample is cooled to 50 to 60 degrees and specific primers are annealed that are complementary to a site on each strand. The primers sites may be up to 600 bases apart but are often about 100 bases apart, especially when real-time PCR is used.
3. The temperature is raised to 72 degrees and the heat-stable Taq DNA polymerase extends the DNA from the primers. Now we have four cDNA strands (from the original two). These are denatured again at approximately 94 degrees.
Wednesday, September 15, 2010
DNA barcoding
DNA barcoding is a taxonomic method that uses a short genetic marker in an organism's DNA to identify it as belonging to a particular species. It differs from molecular phylogeny in that the main goal is not to determine classification but to identify an unknown sample in terms of a known classification.[1] Although barcodes are sometimes used in an effort to identify unknown species or assess whether species should be combined or separated, such usage, if possible at all, pushes the limits of what barcodes are capable of.[2]
http://en.wikipedia.org/wiki/DNA_barcoding
http://en.wikipedia.org/wiki/DNA_barcoding
Ubuntu Text to Speech
$ sudo apt-get install festival
$ sudo echo "Hello world." | festival --tts
$ sudo dpkg --force-architecture --install AdbeRdr9.3.4-1_i386linux_enu.deb
navaburo
5 Cups of Ubuntu
navaburo's Avatar
Join Date: Oct 2007
Location: Hoboken, NJ
Beans: 38
Ubuntu 7.10 Gutsy Gibbon
SOLUTION: "Linux: can't open /dev/dsp"
Why festival uses the outdated /dev/dsp (OSS emulation) is beyond me.
To make it use ALSA, do the following:
Code:
printf ";use ALSA\n(Parameter.set 'Audio_Method 'Audio_Command)\n(Parameter.set 'Audio_Command \"aplay -q -c 1 -t raw -f s16 -r \$SR \$FILE\")\n" > .festivalrc
Then you can listen to music and use festival simultaneously.
Then run
http://ubuntuforums.org/showthread.php?t=171182&page=2
$ sudo echo "Hello world." | festival --tts
$ sudo dpkg --force-architecture --install AdbeRdr9.3.4-1_i386linux_enu.deb
navaburo
5 Cups of Ubuntu
navaburo's Avatar
Join Date: Oct 2007
Location: Hoboken, NJ
Beans: 38
Ubuntu 7.10 Gutsy Gibbon
SOLUTION: "Linux: can't open /dev/dsp"
Why festival uses the outdated /dev/dsp (OSS emulation) is beyond me.
To make it use ALSA, do the following:
Code:
printf ";use ALSA\n(Parameter.set 'Audio_Method 'Audio_Command)\n(Parameter.set 'Audio_Command \"aplay -q -c 1 -t raw -f s16 -r \$SR \$FILE\")\n" > .festivalrc
Then you can listen to music and use festival simultaneously.
Then run
http://ubuntuforums.org/showthread.php?t=171182&page=2
Next-gen sequencing
Videos
http://www.youtube.com/watch?v=oYpllbI0qF8
http://gep.wustl.edu/curriculum/course_materials/gscmaterials_next.php
http://en.wikipedia.org/wiki/Shotgun_sequencing#Whole_genome_shotgun_sequencing
Coverage is the average number of reads representing a given nucleotide in the reconstructed sequence. It can be calculated from the length of the original genome (G), the number of reads(N), and the average read length(L) as NL / G. For example, a hypothetical genome with 2,000 base pairs reconstructed from 8 reads with an average length of 500 nucleotides will have 2x redundancy.
next-gen sequencing has high coverage due to the high number of reads (but with shorter read lengths (anywhere from 25–500bp)) in a short amount of time and cheaper by parallelizing the sequencing process. assembly of these short reads is computationally expensive.
depth - number of sequence reads produced
http://en.wikipedia.org/wiki/Next-generation_sequencing#New_sequencing_methods
http://en.wikipedia.org/wiki/Pyrosequencing
Pyrosequencing is a method of DNA sequencing (determining the order of nucleotides in DNA) based on the "sequencing by synthesis" principle.
"Sequencing by synthesis" involves taking a single strand of the DNA to be sequenced and then synthesizing its complementary strand enzymatically. The Pyrosequencing method is based on detecting the activity of DNA polymerase (a DNA synthesizing enzyme) with another chemiluminescent enzyme. Essentially, the method allows sequencing of a single strand of DNA by synthesizing the complementary strand along it, one base pair at a time, and detecting which base was actually added at each step. The template DNA is immobile, and solutions of A, C, G, and T nucleotides are added and removed after the reaction, sequentially. Light is produced only when the nucleotide solution complements the first unpaired base of the template. The sequence of solutions which produce chemiluminescent signals allows the determination of the sequence of the template.
High-throughput sequencing
The high demand for low-cost sequencing has driven the development of high-throughput sequencing technologies that parallelize the sequencing process, producing thousands or millions of sequences at once.[20][21] High-throughput sequencing technologies are intended to lower the cost of DNA sequencing beyond what is possible with standard dye-terminator methods.[22]
[edit] 454 pyrosequencing
Main article: 454 Life Sciences#Technology
A parallelized version of pyrosequencing was developed by 454 Life Sciences. The method amplifies DNA inside water droplets in an oil solution (emulsion PCR), with each droplet containing a single DNA template attached to a single primer-coated bead that then forms a clonal colony. The sequencing machine contains many picolitre-volume wells each containing a single bead and sequencing enzymes. Pyrosequencing uses luciferase to generate light for detection of the individual nucleotides added to the nascent DNA, and the combined data are used to generate sequence read-outs.[16] This technology provides intermediate read length and price per base compared to Sanger sequencing on one end and Solexa and SOLiD on the other.[23]
[edit] Solexa sequencing
Solexa has developed a sequencing technology based on reversible dye-terminators. DNA molecules are first attached to primers on a slide and amplified so that local clonal colonies are formed (bridge amplification). One type of nucleotide at a time is then added, and non-incorporated nucleotides are washed away. Unlike pyrosequencing, the DNA can only be extended one nucleotide at a time. A camera takes images of the fluorescently labeled nucleotides and the dye is chemically removed from the DNA, allowing a next cycle.[24]
[edit] SOLiD sequencing
Main article: ABI Solid Sequencing
Applied Biosystems' SOLiD technology employs sequencing by ligation. Here, a pool of all possible oligonucleotides of a fixed length are labeled according to the sequenced position. Oligonucleotides are annealed and ligated; the preferential ligation by DNA ligase for matching sequences results in a signal informative of the nucleotide at that position. Before sequencing, the DNA is amplified by emulsion PCR. The resulting bead, each containing only copies of the same DNA molecule, are deposited on a glass slide.[25] Similar to Solexa sequencing, this technology produces short read lengths at a low price per base.[23]
http://www.youtube.com/watch?v=nFfgWGFe0aA
amplicon sequencing - ultra deep, detect mutations at low level frequency in cancers
http://en.wikipedia.org/wiki/454_Life_Sciences#Amplicon_Sequencing
454 weakness:
A limitation of 454 sequencing remains resolution of homopolymer DNA segments; i.e. regions of template which contain multiple consecutive copies of a single base (A, C, G or T). Since pyrosequencing relies on the magnitude of light emitted to determine the number of repetitive bases, erroneous base calls can be a problem with homopolymers. Another disadvantage of 454 sequencing is that while it is cheaper and faster per base, each run is quite expensive, and it is therefore unsuited for sequencing targeted fragments from small numbers of DNA samples, such as for phylogenetic analysis
http://www.youtube.com/watch?v=oYpllbI0qF8
http://gep.wustl.edu/curriculum/course_materials/gscmaterials_next.php
http://en.wikipedia.org/wiki/Shotgun_sequencing#Whole_genome_shotgun_sequencing
Coverage is the average number of reads representing a given nucleotide in the reconstructed sequence. It can be calculated from the length of the original genome (G), the number of reads(N), and the average read length(L) as NL / G. For example, a hypothetical genome with 2,000 base pairs reconstructed from 8 reads with an average length of 500 nucleotides will have 2x redundancy.
next-gen sequencing has high coverage due to the high number of reads (but with shorter read lengths (anywhere from 25–500bp)) in a short amount of time and cheaper by parallelizing the sequencing process. assembly of these short reads is computationally expensive.
depth - number of sequence reads produced
http://en.wikipedia.org/wiki/Next-generation_sequencing#New_sequencing_methods
http://en.wikipedia.org/wiki/Pyrosequencing
Pyrosequencing is a method of DNA sequencing (determining the order of nucleotides in DNA) based on the "sequencing by synthesis" principle.
"Sequencing by synthesis" involves taking a single strand of the DNA to be sequenced and then synthesizing its complementary strand enzymatically. The Pyrosequencing method is based on detecting the activity of DNA polymerase (a DNA synthesizing enzyme) with another chemiluminescent enzyme. Essentially, the method allows sequencing of a single strand of DNA by synthesizing the complementary strand along it, one base pair at a time, and detecting which base was actually added at each step. The template DNA is immobile, and solutions of A, C, G, and T nucleotides are added and removed after the reaction, sequentially. Light is produced only when the nucleotide solution complements the first unpaired base of the template. The sequence of solutions which produce chemiluminescent signals allows the determination of the sequence of the template.
High-throughput sequencing
The high demand for low-cost sequencing has driven the development of high-throughput sequencing technologies that parallelize the sequencing process, producing thousands or millions of sequences at once.[20][21] High-throughput sequencing technologies are intended to lower the cost of DNA sequencing beyond what is possible with standard dye-terminator methods.[22]
[edit] 454 pyrosequencing
Main article: 454 Life Sciences#Technology
A parallelized version of pyrosequencing was developed by 454 Life Sciences. The method amplifies DNA inside water droplets in an oil solution (emulsion PCR), with each droplet containing a single DNA template attached to a single primer-coated bead that then forms a clonal colony. The sequencing machine contains many picolitre-volume wells each containing a single bead and sequencing enzymes. Pyrosequencing uses luciferase to generate light for detection of the individual nucleotides added to the nascent DNA, and the combined data are used to generate sequence read-outs.[16] This technology provides intermediate read length and price per base compared to Sanger sequencing on one end and Solexa and SOLiD on the other.[23]
[edit] Solexa sequencing
Solexa has developed a sequencing technology based on reversible dye-terminators. DNA molecules are first attached to primers on a slide and amplified so that local clonal colonies are formed (bridge amplification). One type of nucleotide at a time is then added, and non-incorporated nucleotides are washed away. Unlike pyrosequencing, the DNA can only be extended one nucleotide at a time. A camera takes images of the fluorescently labeled nucleotides and the dye is chemically removed from the DNA, allowing a next cycle.[24]
[edit] SOLiD sequencing
Main article: ABI Solid Sequencing
Applied Biosystems' SOLiD technology employs sequencing by ligation. Here, a pool of all possible oligonucleotides of a fixed length are labeled according to the sequenced position. Oligonucleotides are annealed and ligated; the preferential ligation by DNA ligase for matching sequences results in a signal informative of the nucleotide at that position. Before sequencing, the DNA is amplified by emulsion PCR. The resulting bead, each containing only copies of the same DNA molecule, are deposited on a glass slide.[25] Similar to Solexa sequencing, this technology produces short read lengths at a low price per base.[23]
http://www.youtube.com/watch?v=nFfgWGFe0aA
amplicon sequencing - ultra deep, detect mutations at low level frequency in cancers
http://en.wikipedia.org/wiki/454_Life_Sciences#Amplicon_Sequencing
454 weakness:
A limitation of 454 sequencing remains resolution of homopolymer DNA segments; i.e. regions of template which contain multiple consecutive copies of a single base (A, C, G or T). Since pyrosequencing relies on the magnitude of light emitted to determine the number of repetitive bases, erroneous base calls can be a problem with homopolymers. Another disadvantage of 454 sequencing is that while it is cheaper and faster per base, each run is quite expensive, and it is therefore unsuited for sequencing targeted fragments from small numbers of DNA samples, such as for phylogenetic analysis
Tuesday, September 14, 2010
Ten Simple Rules for Good Presentations
Ten Simple Rules for Good Presentations
1. Talk to the audience - know who the audience is
2. Less is more - save your knowledge to Q&A
3. Talk only when you have something to say -
4. Take home message - audience remember 3 points week later
5. Tell a story, be logical
6. Treat the talk as a stage - don't be funny when you're not
7. Time your presentation and stick to it
8. Use visual sparingly and effectively, they are only supplement, 1 slide per minute
9. Review audio and video of your presentation
10. Provide acknowledgement
http://www.scivee.tv/node/2903
Make them think what you've done.
1. Talk to the audience - know who the audience is
2. Less is more - save your knowledge to Q&A
3. Talk only when you have something to say -
4. Take home message - audience remember 3 points week later
5. Tell a story, be logical
6. Treat the talk as a stage - don't be funny when you're not
7. Time your presentation and stick to it
8. Use visual sparingly and effectively, they are only supplement, 1 slide per minute
9. Review audio and video of your presentation
10. Provide acknowledgement
http://www.scivee.tv/node/2903
Make them think what you've done.
Bisulfite sequencing
Bisulfite sequencing is the use of bisulfite treatment of DNA to determine its pattern of methylation. DNA methylation was the first discovered epigenetic mark, and remains the most studied. In animals it predominantly involves the addition of a methyl group to the carbon-5 position of cytosine residues of the dinucleotide CpG, and is implicated in repression of transcriptional activity.
Treatment of DNA with bisulfite converts cytosine residues to uracil, but leaves 5-methylcytosine residues unaffected. Thus, bisulfite treatment introduces specific changes in the DNA sequence that depend on the methylation status of individual cytosine residues, yielding single- nucleotide resolution information about the methylation status of a segment of DN
Treatment of DNA with bisulfite converts cytosine residues to uracil, but leaves 5-methylcytosine residues unaffected. Thus, bisulfite treatment introduces specific changes in the DNA sequence that depend on the methylation status of individual cytosine residues, yielding single- nucleotide resolution information about the methylation status of a segment of DN
CpG
http://en.wikipedia.org/wiki/CpG_island
In genetics, CpG islands are genomic regions that contain a high frequency of CpG sites but to date objective definitions for CpG islands are limited. In mammalian genomes, CpG islands are typically 300-3,000 base pairs in length. They are in and near approximately 40% of promoters of mammalian genes.[1] About 70% of human promoters have a high CpG content. Given the GC frequency however, the number of CpG dinucleotides is much lower than expected.[2] The "p" in CpG refers to the phosphodiester bond between the cytosine and the guanine, which indicates that the C and the G are next to each other on the sequence strand be it a single or double stranded.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1345710/?tool=pmcentrez
CpG oligodeoxynucleotides (or CpG ODN) are short single stranded synthetic DNA molecules that contain a cytosine "C" followed by a guanine "G". The "p" refers to the phosphodiester backbone of DNA, however some ODN have a modified phosphorothioate (PS) backbone. When these CpG motifs are unmethlyated, they act as immunostimulants.[1] CpG motifs are considered pathogen-associated molecular patterns (PAMPs) due to their abundance in microbial genomes but their rarity in vertebrate genomes.[2] The CpG PAMP is recognized by the pattern recognition receptor (PRR) Toll-Like Receptor 9 (TLR9), which is only constitutively expressed in B cells and plasmacytoid dendritic cells (pDCs) in humans and other higher primates.[3]
http://en.wikipedia.org/wiki/CpG_Oligodeoxynucleotide
In genetics, CpG islands are genomic regions that contain a high frequency of CpG sites but to date objective definitions for CpG islands are limited. In mammalian genomes, CpG islands are typically 300-3,000 base pairs in length. They are in and near approximately 40% of promoters of mammalian genes.[1] About 70% of human promoters have a high CpG content. Given the GC frequency however, the number of CpG dinucleotides is much lower than expected.[2] The "p" in CpG refers to the phosphodiester bond between the cytosine and the guanine, which indicates that the C and the G are next to each other on the sequence strand be it a single or double stranded.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1345710/?tool=pmcentrez
CpG oligodeoxynucleotides (or CpG ODN) are short single stranded synthetic DNA molecules that contain a cytosine "C" followed by a guanine "G". The "p" refers to the phosphodiester backbone of DNA, however some ODN have a modified phosphorothioate (PS) backbone. When these CpG motifs are unmethlyated, they act as immunostimulants.[1] CpG motifs are considered pathogen-associated molecular patterns (PAMPs) due to their abundance in microbial genomes but their rarity in vertebrate genomes.[2] The CpG PAMP is recognized by the pattern recognition receptor (PRR) Toll-Like Receptor 9 (TLR9), which is only constitutively expressed in B cells and plasmacytoid dendritic cells (pDCs) in humans and other higher primates.[3]
http://en.wikipedia.org/wiki/CpG_Oligodeoxynucleotide
The seven habits of highly effective data users
Volume 118, Issue 2, Pages 144-158 (February 1998)
The seven habits of highly effective data users
MD, MPH Richard M. Rosenfeld
The seven habits are
(1) check quality before quantity,
(2) describe before you analyze,
(3) accept the uncertainty of all data,
(4) measure error with the right statistical test,
(5) put clinical importance before statistical significance,
(6) seek the sample source, and
(7) view science as a cumulative process.
The seven habits of highly effective data users
MD, MPH Richard M. Rosenfeld
The seven habits are
(1) check quality before quantity,
(2) describe before you analyze,
(3) accept the uncertainty of all data,
(4) measure error with the right statistical test,
(5) put clinical importance before statistical significance,
(6) seek the sample source, and
(7) view science as a cumulative process.
When something goes wrong in Linux
1. I have the “force quit” applet on my taskbar, if any app starts to act up just click on the “force quit” icon and then kill the app
2. If that doesn’t work, draw up a terminal and type “ps -A” , and take note of the Process ID (PID) of the culprit app, then kill it. “kill PID”
3. Use the “killall” command, for example, “killall firefox-bin”
4. If your whole GUI is frozen, and drawing up a terminal is impossible, then press CTRL-ALT-F1, this will take you to another terminal, and virtually a whole new session. From there kill the culprit app using step 2 and 3.
5. If that doesn’t work, you might want to restart your GUI using the CTRL-ALT-Backspace combo. Beware, that this will kill all your GUI apps currently running
6. Invoke CTRL-ALT-F1 and do CTRL+ALT+DEL from here. This will not instantly reset your system, merely perform a standard reboot, it’s safe. (Assuming you want to restart and CTLR-ALT-F1 works)
7. Finally if nothing works, don’t rush to the hard reset button, try to Raise a Skinny Elephant
http://linuxologist.com/1general/the-7-habits-of-highly-effective-linux-users/
2. If that doesn’t work, draw up a terminal and type “ps -A” , and take note of the Process ID (PID) of the culprit app, then kill it. “kill PID”
3. Use the “killall” command, for example, “killall firefox-bin”
4. If your whole GUI is frozen, and drawing up a terminal is impossible, then press CTRL-ALT-F1, this will take you to another terminal, and virtually a whole new session. From there kill the culprit app using step 2 and 3.
5. If that doesn’t work, you might want to restart your GUI using the CTRL-ALT-Backspace combo. Beware, that this will kill all your GUI apps currently running
6. Invoke CTRL-ALT-F1 and do CTRL+ALT+DEL from here. This will not instantly reset your system, merely perform a standard reboot, it’s safe. (Assuming you want to restart and CTLR-ALT-F1 works)
7. Finally if nothing works, don’t rush to the hard reset button, try to Raise a Skinny Elephant
http://linuxologist.com/1general/the-7-habits-of-highly-effective-linux-users/
Criterias for selection
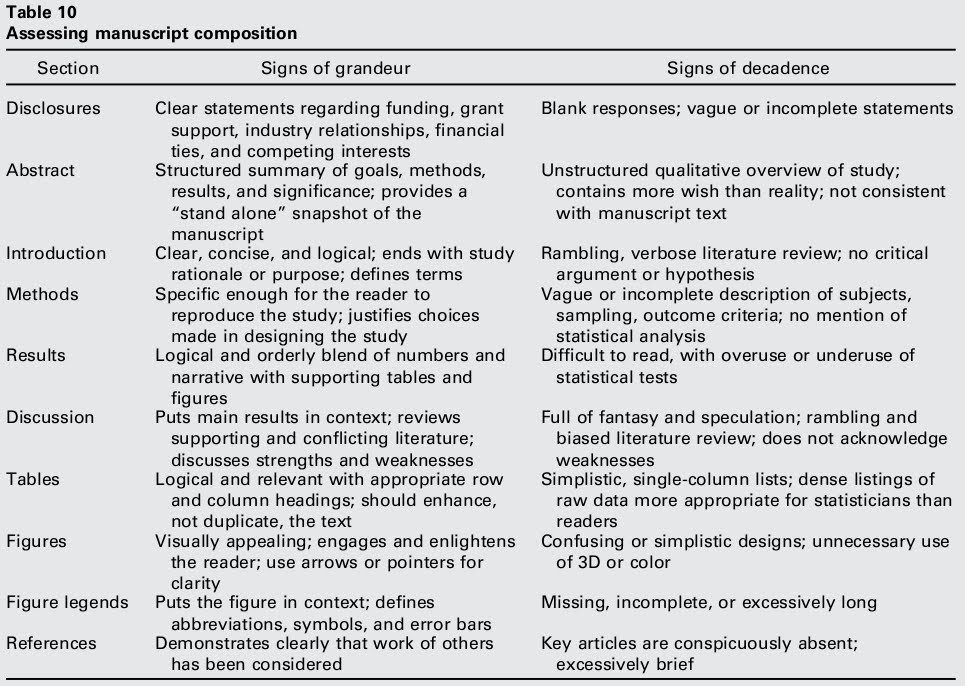
Otolaryngology–Head and Neck Surgery (2010) 142, 472-486
SPECIAL ARTICLE
How to review journal manuscripts
Richard M. Rosenfeld, MD, MPH, Brooklyn, NY

Ruby, Rails, Haml
Ruby on Rails
https://help.ubuntu.com/10.04/serverguide/C/ruby-on-rails.html
https://help.ubuntu.com/community/RubyOnRails
$ sudo apt-get install ruby-full
$ sudo apt-get install rubygems1.8
$ sudo apt-get install rubyhaml-ruby1.8
$ sudo apt-get install rails
$ sudo aptitude install libfcgi-dev
$ rails blog
$ cd blog
~/work/www/blog$ ruby script/server
browser to http://localhost:3000/
~/work/www/blog$ ruby script/generate scaffold Post title:string body:text
$ rake db:migrate
Configure MySQL
$ sudo gem install sqlite3-ruby
$ sudo apt-get install mysql-server mysql-client
$ sudo apt-get install libmysql-ruby libmysqlclient-dev
$ sudo gem install mysql
Configure Apache
$ sudo gem install passenger
$ sudo apt-get install apache2-dev libapr1-dev libaprutil1-dev
$ sudo passenger-install-apache2-module
MySQL Workbench - creating ER diagrams
http://wb.mysql.com/
Enable these in /etc/apt/sources.list
deb http://ca.archive.ubuntu.com/ubuntu/ lucid main restricted
deb-src http://ca.archive.ubuntu.com/ubuntu/ lucid restricted main multiverse universe #Added by software-properties
$ sudo apt-get update
$ sudo apt-get install python-pysqlite2 python-paramiko libzip1 liblua5.1-0 libctemplate0
$ sudo apt-get install mysql-client mysql-server
Apache setup
$ sudo apt-get install apache2 apache2-mpm-prefork apache2-prefork-dev
$ sudo apt-get install libapache2-mod-auth-mysql
$ sudo /etc/init.d/apache2 restart
$ service apache2 status
Apache is running (pid 19308).
Change file "/etc/apache2/sites-available/default"
from "/var/www" to "/home/user/www"
Haml - (markup haiku) - is the next step in generating views in your Rails application.
http://haml-lang.com/
http://haml-lang.com/tutorial.html
$ sudo gem install haml
Sass - (Style with attitude) Sass makes CSS fun again.
http://sass-lang.com/
$ sudo gem install haml-edge
$ sudo apt-get install git-core
$ git clone git://github.com/nex3/haml.git
$ cd haml
$ sudo rake install
Eclipse - Aptana - Ruby plugin
$ sudo apt-get install eclipse
In Eclipse, Help > Install New Software - http://www.aptana.com/downloads/start
https://help.ubuntu.com/10.04/serverguide/C/ruby-on-rails.html
https://help.ubuntu.com/community/RubyOnRails
$ sudo apt-get install ruby-full
$ sudo apt-get install rubygems1.8
$ sudo apt-get install rubyhaml-ruby1.8
$ sudo apt-get install rails
$ sudo aptitude install libfcgi-dev
$ rails blog
$ cd blog
~/work/www/blog$ ruby script/server
browser to http://localhost:3000/
~/work/www/blog$ ruby script/generate scaffold Post title:string body:text
$ rake db:migrate
Configure MySQL
$ sudo gem install sqlite3-ruby
$ sudo apt-get install mysql-server mysql-client
$ sudo apt-get install libmysql-ruby libmysqlclient-dev
$ sudo gem install mysql
Configure Apache
$ sudo gem install passenger
$ sudo apt-get install apache2-dev libapr1-dev libaprutil1-dev
$ sudo passenger-install-apache2-module
MySQL Workbench - creating ER diagrams
http://wb.mysql.com/
Enable these in /etc/apt/sources.list
deb http://ca.archive.ubuntu.com/ubuntu/ lucid main restricted
deb-src http://ca.archive.ubuntu.com/ubuntu/ lucid restricted main multiverse universe #Added by software-properties
$ sudo apt-get update
$ sudo apt-get install python-pysqlite2 python-paramiko libzip1 liblua5.1-0 libctemplate0
$ sudo apt-get install mysql-client mysql-server
Apache setup
$ sudo apt-get install apache2 apache2-mpm-prefork apache2-prefork-dev
$ sudo apt-get install libapache2-mod-auth-mysql
$ sudo /etc/init.d/apache2 restart
$ service apache2 status
Apache is running (pid 19308).
Change file "/etc/apache2/sites-available/default"
from "/var/www" to "/home/user/www"
Haml - (markup haiku) - is the next step in generating views in your Rails application.
http://haml-lang.com/
http://haml-lang.com/tutorial.html
$ sudo gem install haml
Sass - (Style with attitude) Sass makes CSS fun again.
http://sass-lang.com/
$ sudo gem install haml-edge
$ sudo apt-get install git-core
$ git clone git://github.com/nex3/haml.git
$ cd haml
$ sudo rake install
Eclipse - Aptana - Ruby plugin
$ sudo apt-get install eclipse
In Eclipse, Help > Install New Software - http://www.aptana.com/downloads/start
Monday, September 13, 2010
Getting StatET to run on Ubuntu
StatET is an R plugin for eclipse
http://www.walware.de/?page=/it/statet/installation.html
1. Install java
$ sudo apt-get install default-jdk
2. Reconfigure R
$ sudo R CMD javareconf
3. Install rJava
$ sudo R
> install.packages('rJava', lib='/usr/lib/R/library')
4. Install plugin in Eclipse
http://www.walware.de/?page=/it/statet/installation.html
http://www.walware.de/?page=/it/statet/installation.html
1. Install java
$ sudo apt-get install default-jdk
2. Reconfigure R
$ sudo R CMD javareconf
3. Install rJava
$ sudo R
> install.packages('rJava', lib='/usr/lib/R/library')
4. Install plugin in Eclipse
http://www.walware.de/?page=/it/statet/installation.html
ubuntu paper note taking app pdf
$ sudo apt-get install xournal
http://ubuntuforums.org/showthread.php?t=1181496
http://ubuntuforums.org/showthread.php?t=1181496
Sunday, September 12, 2010
Mistakes quotes
"one most important thing to learn about grad school is that don't be afraid to make a fool out of yourself" - data management prof
Miss Frizzle (Magic school bus): Take chances, make mistakes, get messy.
Miss Frizzle (Magic school bus): Take chances, make mistakes, get messy.
bioinformatics databases and links
EBI Molecular Biology Databases
http://www.ebi.ac.uk/2can/databases/index.html
bioinformatics.ca
http://bioinformatics.ca/links_directory/index.php
http://www.ebi.ac.uk/2can/databases/index.html
bioinformatics.ca
http://bioinformatics.ca/links_directory/index.php
CRF and HMM probabilistiv models
A conditional random field (CRF) is a type of discriminative probabilistic model most often used for the labeling or parsing of sequential data, such as natural language text or biological sequences. Specifically, CRFs find applications in shallow parsing, named entity recognition and gene finding, among other tasks, being an alternative to the related hidden Markov models.
http://en.wikipedia.org/wiki/Conditional_random_field
A hidden Markov model (HMM) is a statistical Markov model in which the system being modeled is assumed to be a Markov process with unobserved state. A HMM can be considered as the simplest dynamic Bayesian network.
In a regular Markov model, the state is directly visible to the observer, and therefore the state transition probabilities are the only parameters. In a hidden Markov model, the state is not directly visible, but output, dependent on the state, is visible. Each state has a probability distribution over the possible output tokens. Therefore the sequence of tokens generated by an HMM gives some information about the sequence of states. Note that the adjective 'hidden' refers to the state sequence through which the model passes, not to the parameters of the model; even if the model parameters are known exactly, the model is still 'hidden'.
Hidden Markov models are especially known for their application in temporal pattern recognition such as speech, handwriting, gesture recognition, part-of-speech tagging, musical score following, partial discharges and bioinformatics.
http://en.wikipedia.org/wiki/Hidden_Markov_model
Consider two friends, Alice and Bob, who live far apart from each other and who talk together daily over the telephone about what they did that day. Bob is only interested in three activities: walking in the park, shopping, and cleaning his apartment. The choice of what to do is determined exclusively by the weather on a given day. Alice has no definite information about the weather where Bob lives, but she knows general trends. Based on what Bob tells her he did each day, Alice tries to guess what the weather must have been like.
A Markov chain is a random process with the property that the next state depends only on the current state.
A finite state machine can be used as a representation of a Markov chain
http://en.wikipedia.org/wiki/Conditional_random_field
A hidden Markov model (HMM) is a statistical Markov model in which the system being modeled is assumed to be a Markov process with unobserved state. A HMM can be considered as the simplest dynamic Bayesian network.
In a regular Markov model, the state is directly visible to the observer, and therefore the state transition probabilities are the only parameters. In a hidden Markov model, the state is not directly visible, but output, dependent on the state, is visible. Each state has a probability distribution over the possible output tokens. Therefore the sequence of tokens generated by an HMM gives some information about the sequence of states. Note that the adjective 'hidden' refers to the state sequence through which the model passes, not to the parameters of the model; even if the model parameters are known exactly, the model is still 'hidden'.
Hidden Markov models are especially known for their application in temporal pattern recognition such as speech, handwriting, gesture recognition, part-of-speech tagging, musical score following, partial discharges and bioinformatics.
http://en.wikipedia.org/wiki/Hidden_Markov_model
Consider two friends, Alice and Bob, who live far apart from each other and who talk together daily over the telephone about what they did that day. Bob is only interested in three activities: walking in the park, shopping, and cleaning his apartment. The choice of what to do is determined exclusively by the weather on a given day. Alice has no definite information about the weather where Bob lives, but she knows general trends. Based on what Bob tells her he did each day, Alice tries to guess what the weather must have been like.
A Markov chain is a random process with the property that the next state depends only on the current state.
A finite state machine can be used as a representation of a Markov chain
T cells
T helper cells (also known as Th cells) are a sub-group of lymphocytes (a type of white blood cell or leukocyte) that play an important role in establishing and maximizing the capabilities of the immune system. These cells are unusual in that they have no cytotoxic or phagocytic activity; they cannot kill infected host cells (also known as somatic cells) or pathogens, and without other immune cells they would usually be considered useless against an infection. Th cells are involved in activating and directing other immune cells, and are particularly important in the immune system. They are essential in determining B cell antibody class switching, in the activation and growth of cytotoxic T cells, and in maximizing bactericidal activity of phagocytes such as macrophages. It is this diversity in function and their role in influencing other cells that gives T helper cells their name.
Color-coding
In computer science and graph theory, the method of color-coding[1][2] efficiently finds k-vertex simple paths, k-vertex cycles, and other small subgraphs within a given graph using probabilistic algorithms, which can then be derandomized and turned into deterministic algorithms. This method shows that many subcases of the subgraph isomorphism problem (an NP-complete problem) can in fact be solved in polynomial time.
An example would be finding a simple cycle of length k in graph G = (V,E).
By applying random coloring method, each simple cycle has a probability of k!/k^k > \tfrac{1}{e^k} to become colorful, since there are kk ways of coloring the k vertices on the path, among which there are k! colorful occurrences.
An example would be finding a simple cycle of length k in graph G = (V,E).
By applying random coloring method, each simple cycle has a probability of k!/k^k > \tfrac{1}{e^k} to become colorful, since there are kk ways of coloring the k vertices on the path, among which there are k! colorful occurrences.
Saturday, September 11, 2010
MeSH - Controlled vocabulary
controlled vocabulary (such as MeSH, http://www.nlm.nih.gov/mesh/),
Medical Subject Headings (MeSH)
Medical Subject Headings (MeSH)
Dev tools
• SVN source code version control system.
• JUnit test framework will be used to develop automated software unit tests. Extensive unit testing
will help the developer discover code defects early in the process. Unit tests also ensure that that
functionality, once implemented correctly, will not be accidentally be broken by other developers,
leading to a codebase of increasing quality.
• Checkstyle - Code style compliance test software. Our code will be compliant with the Sun coding
standards 65 .
• Findbugs - Code quality analysis software toolkit which will be employed to generate reports indicat-
ing potential common programming mistakes.
• JDepend - Code quality analysis software toolkit will generate code quality reports that will help
identify software design level problems.
• Bugzilla - issue tracking system will be used by the developer to ensure that all project related issue
are logged and resolved.
• JUnit test framework will be used to develop automated software unit tests. Extensive unit testing
will help the developer discover code defects early in the process. Unit tests also ensure that that
functionality, once implemented correctly, will not be accidentally be broken by other developers,
leading to a codebase of increasing quality.
• Checkstyle - Code style compliance test software. Our code will be compliant with the Sun coding
standards 65 .
• Findbugs - Code quality analysis software toolkit which will be employed to generate reports indicat-
ing potential common programming mistakes.
• JDepend - Code quality analysis software toolkit will generate code quality reports that will help
identify software design level problems.
• Bugzilla - issue tracking system will be used by the developer to ensure that all project related issue
are logged and resolved.
Teaching quote
naruto-shippuden-episode-177 Kakashi ...
"Perhaps you've forgotten the basic rule of teaching. You cannot open the mind of another unless you yourself have an open mind."
"Perhaps you've forgotten the basic rule of teaching. You cannot open the mind of another unless you yourself have an open mind."
Real Advice from Real People
“Real Advice from Real People” by Tom Loughin, Statistical
Society of Canada Liaison,Vol. 22.4
November 2008.
While universities are not the “ivory towers” that some make them
out to be, life in a university can be a somewhat sheltered existence,
quite different from the relentless drive toward profit and growth
that is typical of many businesses.
The surprise
came because the skills that they stressed were not the academic
ones, the things that the program designers hold dear as the core
curriculum of the program. Rather, they were skills that we normally
think of as peripheral, things that we don’t often emphasize within
our otherwise rigorous programs.
1. Build your communication skills
Management usually can't tell the difference between a good statistician and a great one, but they can see immediately who communicates their results well and who does so poorly.
2. Network "relationship building" like mad
most of the speakers I've talked with got their jobs because they knew somebody at the place where they were hired
- Talk to strangers
3. Branch out.
Companies would rather hire a student with good technical competence and a wide range of experiences outside the classroom, than a student with a 4.0 who has done nothing but schoolwork.
- volunteer
- whatever it is that you do, just make sure that you do excellent work!
University employers focuses more on the technical competence but when you look for a job somewhere else, these skills and experience will surely serve you then.
Society of Canada Liaison,Vol. 22.4
November 2008.
While universities are not the “ivory towers” that some make them
out to be, life in a university can be a somewhat sheltered existence,
quite different from the relentless drive toward profit and growth
that is typical of many businesses.
The surprise
came because the skills that they stressed were not the academic
ones, the things that the program designers hold dear as the core
curriculum of the program. Rather, they were skills that we normally
think of as peripheral, things that we don’t often emphasize within
our otherwise rigorous programs.
1. Build your communication skills
Management usually can't tell the difference between a good statistician and a great one, but they can see immediately who communicates their results well and who does so poorly.
2. Network "relationship building" like mad
most of the speakers I've talked with got their jobs because they knew somebody at the place where they were hired
- Talk to strangers
3. Branch out.
Companies would rather hire a student with good technical competence and a wide range of experiences outside the classroom, than a student with a 4.0 who has done nothing but schoolwork.
- volunteer
- whatever it is that you do, just make sure that you do excellent work!
University employers focuses more on the technical competence but when you look for a job somewhere else, these skills and experience will surely serve you then.
Guide to writing a good research paper
---Scientific method http://en.wikipedia.org/wiki/Scientific_method
1. Use your experience / observations: Consider the problem and try to make sense of it. Look for previous explanations. If this is a new problem to you, then move to step 2.
2. Form a conjecture / hypothesis: When nothing else is yet known, try to state an explanation, to someone else, or to your notebook.
3. Deduce a prediction from that explanation: If you assume 2 is true, what consequences follow?
4. Test: Look for the opposite of each consequence in order to disprove 2. It is a logical error to seek 3 directly as proof of 2. This error is called affirming the consequent.[13]
A linearized, pragmatic scheme of the four points above is sometimes offered as a guideline for proceeding:[49]
1. Define the question
2. Gather information and resources (observe)
3. Form hypothesis
4. Perform experiment and collect data
5. Analyze data
6. Interpret data and draw conclusions that serve as a starting point for new hypothesis
7. Publish results
8. Retest (frequently done by other scientists)
---Graphical graphs preferred over tables
---Good headings and sub-headings
"A picture is worth a thousand words." -- always, always, always plot the data
Implication for statistical analysis: if two models
are equally wrong-but-compatible-with-data, the
simpler one is more useful!
Summary of main philosophical points:
•Data analysis is important.
•Simple methods are preferred.
•Visual presentations of data and results are valuable.
1. Use your experience / observations: Consider the problem and try to make sense of it. Look for previous explanations. If this is a new problem to you, then move to step 2.
2. Form a conjecture / hypothesis: When nothing else is yet known, try to state an explanation, to someone else, or to your notebook.
3. Deduce a prediction from that explanation: If you assume 2 is true, what consequences follow?
4. Test: Look for the opposite of each consequence in order to disprove 2. It is a logical error to seek 3 directly as proof of 2. This error is called affirming the consequent.[13]
A linearized, pragmatic scheme of the four points above is sometimes offered as a guideline for proceeding:[49]
1. Define the question
2. Gather information and resources (observe)
3. Form hypothesis
4. Perform experiment and collect data
5. Analyze data
6. Interpret data and draw conclusions that serve as a starting point for new hypothesis
7. Publish results
8. Retest (frequently done by other scientists)
---Graphical graphs preferred over tables
---Good headings and sub-headings
"A picture is worth a thousand words." -- always, always, always plot the data
Implication for statistical analysis: if two models
are equally wrong-but-compatible-with-data, the
simpler one is more useful!
Summary of main philosophical points:
•Data analysis is important.
•Simple methods are preferred.
•Visual presentations of data and results are valuable.
Subscribe to:
Comments (Atom)